Szervezetünk fehérjéi számos kismolekula kötésére képesek. Ide tartoznak a jelátvivő anyagok, bomlástermékek, antigének vagy éppen a gyógyszermolekulák. Az egyes fehérjéken található kötőhelyek feltérképezése lehetőséget ad arra, hogy megismerjük bennük a kismolekulák elhelyezkedését, sőt újabbakat, például új típusú gyógyszerjelölt molekulákat fejlesszünk, amelyek egy kívánt kötőhelyen hatnak.
A kismolekulák illeszkedéséről akkor kapjuk a leghitelesebb képet, ha a fehérjéről és ligandumáról rendelkezésre áll röntgendiffrakcióval vagy NMR-méréssel meghatározott szerkezet. (NMR: mágneses magrezonancia spektroszkópia). Ezeknek a méréseknek az eredményeiből az egyes atomok koordinátáit kapjuk meg, így felrajzolható a teljes fehérje a beleilleszkedő ligandummal együtt.

1. ábra. Ligandum dokkolása egy fehérjéhez
Ha a fehérjéről csak önmagában áll rendelkezésre háromdimenziós szerkezet, akkor a hozzá kötődő kisméretű molekula helyét ún. dokkolási eljárással ismerhetjük meg (1. ábra). A dokkoló programok legegyszerűbb esetben a fehérje és a kismolekula között fellépő elektrosztatikus és van der Waals-kölcsönhatásokat, illetve hidrogénkötéseket veszik figyelembe. Ezek alapján több lehetőséget kipróbálnak, és a legkedvezőbbeket adják vissza. Az illeszkedés jóságát úgy ellenőrizhetjük, ha a fehérje-ligandum együttes röntgenszerkezetéből kivágjuk a ligandumot, majd visszaillesztjük a fehérje kötőhelyébe. Ha ebben az eljárásban a ligandum vis.szatalál eredeti helyére, akkor bízhatunk abban, hogy az általunk módosított, tervezett vagy egy adatbázisból nyert újabb vegyület illeszkedése is valósághű.

2. ábra. A ligandum elhelyezése
a rácsban (https://www.csb.yale.edu/userguides/datamanip/
autodock/html/Using_AutoDock_305.9.html)
A dokkoláshoz számos kereskedelmi program áll rendelkezésre. Az egyik legelterjedtebb az Autodock, amely ingyenesen letölthető és futtatható akár otthon is. Az Autodock-program a fehérjét vagy egy részét egy ráccsal helyettesíti, amelynek rácspontjaiban kiszámítja a képzeletbeli atomra ható erőteret, majd ezt elvégzi a ligandum összes atomtípusára (2. ábra). Tehát kiszámítja, milyen teret érezne egy nitrogénatom, oxigénatom, vagy kénatom egy adott pontban. A program a ligandumot többféleképpen helyezi el, majd a kapott illeszkedéseket pontozza aszerint, mennyi kedvező kölcsönhatást képes kialakítani a ligandum. Az így kapott értéket „kötési energiaként” adja vis.sza, azaz minél alacsonyabb negatív értéket kapunk, annál kedvezőbb az illeszkedés (3. ábra). Természetesen, az így kapott érték csak közelítés, a tényleges kötési energia értékét nem kapjuk meg, de a dokkolás jó orientációt jelenthet a ligandum kedvező illeszkedésére, illetve az egyes ligandumok közötti rangsor felállítására.
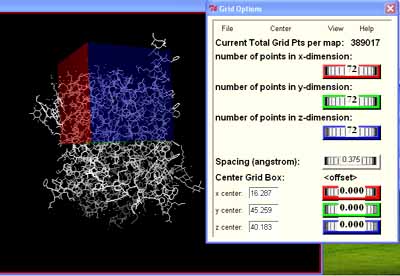
3. ábra. Az Autodock-program felhasználói felülete. A fehérje pálcikaábrázolásban látható, a kékkel jelzett dobozban keressük a ligandum helyét, amelynek méretét a jobb oldali dobozban állítjuk be a középpont és a méret feltüntetésével. Ugyanitt állítjuk be a rácstávolságot is angströmben (spacing)
Egy másik, kedvelt, bár fizetős program a GOLD (4. ábra), amelynek hangzatos neve rövidítést takar (Genetic Optimisation for Ligand Docking). A genetikus algoritmus kifejezés arra utal, hogy a molekula egyes konformereit „kromoszómák” írják le, amelyben a gének jelentik a molekula transzlációs és rotációs, valamint belső (torziós) szabadsági fokait. Ezek keresztezésével számítják ki egy új konformáció kötődési szabadentalpiáját. A GOLD-program használata egyszerű, a fehérje és a ligandum megadásán kívül a kötőhely középpontját kell megadnunk, vagy azokat az aminosavakat, amelyek a kötőhelyet képezik.

4. ábra. A GOLD dokkoló program menüje. Az ábrán látható példán a human GABA transzporter (hGAT-1) fehérjéhez 21 ligandumot dokkolunk ugyanabba a kötőhelybe. A kapott találatokat a GOLDScore pontozófüggvénnyel értékeljük
Hogy néz ki az eredmény?
Az egyik első gyógyszer, amely számítógépes tervezéssel készült, egy rákellenes szer, a Bcr Abl kinázon ható imatinib (Gleevec) volt. Ennek a példáján nézzünk meg egy illeszkedést. Az imatinib dokkolását bármelyik programmal kipróbálhatjuk és ellenőrizhetjük, hiszen rendelkezésre áll a Bcr-Abl kináz röntgendiffrakcióval meghatározott szerkezete és az imatinibé is, valamint a kettő együtt. Ha „kivesszük” az imatinib-molekulát a fehérjéből, majd visszadokkoljuk, akkor az 5. ábrán láthatjuk, mennyire ad pontos illeszkedést a dokkolás a GOLD-programmal.

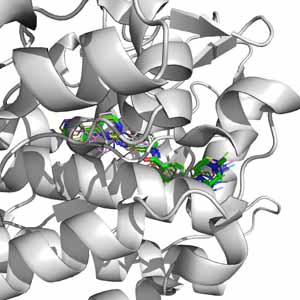
5. ábra. Fönt: az imatinib rákellenes gyógyszer a bcr-abl kináz kötőhelyében (zöld váz – röntgenszerkezeti kép). Lent: színessel a számítógéppel dokkolt találatok. Látható, hogy a dokkolás jól eltalálja az imatinib kötőhelyét a fehérjén
A kinázok vízoldható fehérjék, tehát nem a sejtmembránban helyezkednek el. Az ilyen fehérjéket bejáratott kísérleti körülmények között jól tudják kristályosítani, ami a röntgendiffrakciós szerkezet-meghatározás előfeltétele. A legtöbb gyógyszermolekula azonban membránba ágyazott fehérjéken hat, amelyekből sokszor nagy erőfeszítések árán sem sikerül kristályt készíteni. Ezek szerkezete többnyire nem ismert, így modelleket kell létrehoznunk. Az emberi fehérjék sokszor jól modellezhetők baktériumból származó, hasonló fehérjékből kiindulva, amelyekről viszont már rendelkezésre áll a röntgendiffrakcióval meghatározott szerkezet. A baktériumok közül néhány ugyanis szélsőséges körülményeket is hajlandó elviselni (ilyenek a hő- és sótűrő baktériumok). Ezeknek az organizmusoknak a fehérjéi a kristályosítás körülményeit is jól bírják. A bakteriális és a humán fehérjék között úgy létesíthetünk megfeleltetést, hogy azokat a szakaszaikat illesztjük egymáshoz, amelyek azonos vagy hasonló aminosavakat tartalmaznak. Ha van egy megfeleltetésünk, akkor az ismert szerkezetre számítógépes modellező programokkal „ráhúzhatjuk” a modellezni kívánt fehérje szekvenciáját. Erre a feladatra számítógépes modellező programok állnak rendelkezésre. Közülük a legismertebb, ingyenesen hozzáférhető program a Modeller vagy Swiss PDB Viewer. Ezek a programok a bakteriális fehérje röntgenszerkezetére mintegy ráhúzzák a modellezendő fehérje szekvenciáját, majd egy rövid minimalizálással eligazítják az egymással ütköző oldalláncokat. Az ütközés abból ered, hogy a modellezni kívánt fehérje hosszabb, vagy rövidebb, mint a referenciafehérje, így egyes szakaszokon betoldások vagy kivágások szükségesek.
Az OTKA-pályázat keretében a központi idegrendszer egyik membránfehérjéjét, a gamma-aminovajsav (GABA) transzporter szerkezetét modelleztük egy hőtűrő baktériumból származó hasonló fehérje szerkezete alapján Modeller-programmal. Jelenleg egy belga kutatócsoport bevonásával folyik az az izgalmas munka, amelynek során potenciális gátlószereket fejlesztünk a GABA transzporterek egyes altípusaira. Mivel a GABA a központi idegrendszer fő, gátló jelátvivő anyaga, a gátlás következtében több GABA marad a szinaptikus résben, így terveink szerint ezek az anyagok potenciális antiepileptikumok is lehetnek.
Az írás az OTKA 102166 számú pályázata alapján készült.
| Természet Világa, | 144. évfolyam, 7. szám,
2013. július
http//www.termvil.hu/ https://www.chemonet.hu/TermVil/ |