Egy anyag – két célpont
Lehetőségek a célzott daganatterápiában
A folyóirat két korábbi számában beszámoltunk már azokról a kutatásainkról, amelyek a daganatok célzott elpusztítására irányulnak. A módszer, amelyet célzott vagy irányított daganatterápiának neveznek, azon alapszik, hogy olyan anyagokkal támadják a ráksejteket, amelyek nagy szelektivitással ismerik fel e beteg sejteket. Ezzel az eljárással az egészséges szövetek megkímélhetőek, csökkentve a terápia káros mellékhatásait és javítva a paciens életminőségét a kezelés alatt. A molekulárisan célzott daganatterápia számos ráktípus kezelését forradalmasította. Természetesen ahhoz, hogy ezt a terápiát hatékonyan lehessen alkalmazni, ismerni kell az adott daganaton azokat a molekulákat, amelyek támadhatók a rák elpusztításának reményében. Ezeket a molekulákat a daganat eltávolítása, vagy biopszia során végzett mintavétel után speciális vizsgálatokkal határozzák meg. Az egyik lehetséges kezelés ezen a területen, amikor a daganatsejtekre specifikus ellenanyagot juttatnak a szervezetbe, és ez a ráksejteken megjelenő antigénekhez kötődve aktiválja az immunrendszert a daganat elpusztítására. Ezt az eljárást egyre szélesebb körben alkalmazzák a klinikai onkológiában és része az úgynevezett személyre szabott terápiának.
A másik lehetőség az, hogy a rák ellen alkalmazott gyógyszereket, amelyek önmagukban nem szelektívek, és így a kemoterápiás alkalmazásuk során számos mellékhatást váltanak ki, irányító molekulákhoz kötjük a szelektivitás fokozása érdekében. Az irányító molekulák olyan vegyületek, amelyek a daganatsejteken nagy mennyiségben megjelenő sejtfelszíni struktúrákhoz (pl. receptorokhoz) kötődnek, majd az így kialakult komplexet a ráksejt bekebelezi. Mivel ezek a receptorok az egészséges szöveteken nem vagy csak lényegesen kisebb mennyiségben fordulnak elő, a rákellenes hatóanyagokat nagy szelektivitással lehet a daganatsejtekbe juttatni. Az így bejutatott vegyületből a szabad hatóanyag vagy annak aktív származéka a lebontó enzimek hatására felszabadul, és így kifejthetik gátló hatásukat a daganat növekedésére. A terápiában ez a módszer lényegesen olcsóbb lenne, mint az ellenanyagok alkalmazása. Az ebbe a csoportba sorolható vegyületek közül néhány már a klinikai kipróbálás fázisában van, és remélhetőleg hamarosan bővíthetik a klinikai onkológia eszköztárát. A téma jelentőségét mutatja, hogy számos kutatócsoport foglalkozik szerte a világon olyan kutatásokkal, amelyek során olyan gyógyszereket fejlesztenek ki az irányított daganatterápia céljából, amelyekben a rákellenes szereket irányító peptidekhez kapcsolják. Az MTA–ELTE Peptidkémiai Kutatócsoportban ilyen típusú gyógyszerjelöltek tervezésével, szintézisével és vizsgálatával foglalkozunk.
Ahogy azt a korábbi cikkünkben bemutattuk, a gyógyszer sejtfelszíni receptorokon keresztül történő célba juttatásának hatékonyságát korlátozhatja az, hogy a receptorok száma limitált. Ezért a vegyület koncentrációjának növelése nem feltétlenül vezet a hatékonyság növekedéséhez. Megoldást jelenthet azonban, ha egy irányító molekulához több hatóanyagot kapcsolunk. Ekkor viszont nagyon körültekintően kell eljárni a molekula tervezésénél, nehogy a több hatóanyag jelenléte gátolja az irányító molekula receptor-felismerését és ahhoz való kötődését. Ezért hatékonyabb megoldás lehet, ha a daganatellenes szereket különböző irányító molekulákhoz kapcsoljuk, amelyek eltérő receptorokat ismernek fel a ráksejteken. Az így előállított vegyületek kombinációban történő alkalmazásával – a kapcsolt hatóanyagoktól függően – a komponensek hatása összeadódhat (additív hatás) vagy még erősíthetik is egymás hatását (szinergista hatás) a daganat elpusztításában. Jelenleg ilyen kombinált kezeléseket próbálunk ki in vitro körülmények között annak érdekében, hogy megfelelő összetételű keverékeket tudjunk előállítani. A kombinált kezelés hatékonysága ellenére felmerül a költségfaktor, mivel ebben az esetben legalább két vegyület együttes alkalmazásáról van szó, ami a terápia költségeit jelentősen növelheti. Tehát az lenne az ideális, ha olyan vegyületet tudnánk alkalmazni, amellyel két különböző, a daganat növekedésében szerepet játszó receptort tudnánk egyidejűleg támadni. Mivel a peptidek nagyon szelektívek, erre viszonylag kevés esély van a peptid alapú hatóanyag-célbajuttatás során. Azonban a természet a kezünkbe adott egy lehetőséget, amely meghatározott aszparagin-glicin-arginin (NGR: Asn-Gly-Arg) tripeptid-szekvenciát tartalmazó peptidekhez köthető. Ennek a lehetőségnek a bemutatását tűztük ki célként írásunkban.
A figyelem akkor fordult e peptidek felé, amikor olyan vegyületeket kerestek, amelyek egy másik tripeptid-szekvenciához (RGD: Arg-Gly-Asp) hasonlóan jól kötődnek olyan receptorokhoz (ún. integrin-receptorokhoz), amelyeknek nagy szerepük van a ráksejtek letapadásában, mozgékonyságában és így az áttétek kialakulásában. Ezek a receptorok nagy mennyiségben jelennek meg az újonnan képződő ereken is, amelyek a daganat vérellátásához szükségesek (tumor vaszkularizáció). Ezzel szemben, a már kialakult ereken a receptorok száma elenyésző. Ezért ezek a receptorok jó támadási pontok lehetnek a célzott daganatterápiában. Megállapították azt is, hogy az integrin-receptorhoz jól kötődő peptidek között nagy számban fordulnak elő az Asn-Gly-Arg tripeptid-szekvenciát tartalmazók. Kísérletekkel ki is választottak ilyen peptideket, melyek jó irányító molekuláknak bizonyultak. Azonban kötődésgátlási vizsgálatokban hamar kiderült, hogy ezek a peptidek nem az inegrin-receptorokat ismerik fel, hanem egy másik receptorhoz (CD13) kapcsolódnak nagy affinitással. Annak a kérdésnek a megválaszolása sem váratott sokáig, hogy akkor mégis miért találták azt, hogy ezek a peptidek hatékonyan kötődnek az integrin-receptorokhoz. Az ok a tripeptid-szekvencia szerkezetében keresendő.
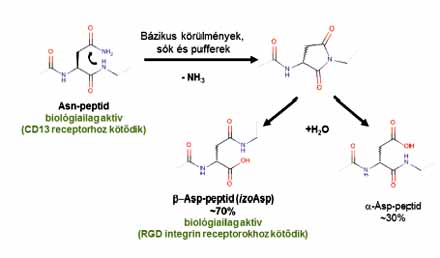
1. ábra. Szukcinimid gyűrűzárás/felnyílás
Azt már korábban is tudták, hogy az aszparaginés glicin(Asn-Gly) egységet tartalmazó peptidek nagyon kön.nyen átalakulnak. Ennek oka az, hogy a glicin aminosavnak nincs oldallánca, így a térgátlás nem akadályozza meg az aszparagin oldalláncának kapcsolódását a glicin N-atomjára ammónia kilépése mellett (1. ábra). A kialakult gyűrűs szerkezet (szukcinimid gyűrű) nem túl stabil, és víz hatására kétféleképpen bomlik fel. A két termék egy aszparaginsavat és glicint (Asp-Gly) tartalmazó egység, illetve egy izo-aszparaginsav– glicin dipeptid egységet tartalmazó származék lesz. Ez utóbbi azt jelenti, hogy a glicin nem az aszparaginsav a-karboxilcsoportjához, hanem az oldalláncában lévő karboxilcsoportjához kapcsolódik. A két termék kb. 30:70 arányban keletkezik (1. ábra).
Ennek a nem enzim által katalizált átalakulásnak a következtében, a nagyobb mennyiségben keletkező izoaszpartilszármazék (izoDGR) az, ami nagy hatékonysággal képes kötődni az integrinreceptorokhoz. Ugyanakkor a másik termék (DGR peptid) nem ismeri fel ezeket a receptorokat.
Vizsgálták azokat a körülményeket, amelyek befolyásolhatják ezt az átalakulást, amit tömegspektrometria segítségével könnyen lehet detektálni az aszparaginsav és az aszparagin közötti 1 egység molekulatömeg különbség alapján. Az oldat pHjának és sókoncentrációjának, valamint hőmérsékletének emelése elősegíti az átalakulást. Természetesen ez jelentősen függ az aszparagin–glicin szekvenciarészletnek a peptidben elfoglalt környezetétől és térszerkezetétől. Egy rendezett szerkezetben lényegesen lassabb, mint egy rendezetlen konformációban. A peptid ciklizálása csökkentheti az átalakulás sebességét az egyenes láncú (lineáris) analóghoz képest.
Néhány kutatócsoport már kifejlesztett és alkalmazott olyan vegyületeket, ahol ezt a tripeptid-szekvenciát tartalmazó peptidet mint irányító molekulát alkalmazták rákellenes szer célzott daganatsejtbe juttatásra. Az esetek többségében azonban nem vizsgálták az irányító molekulaként szolgáló peptidek stabilitását. Zou és munkatársai 2012-ben vetették fel először azt az elméletet, hogy megfelelően megválasztott AsnGly-Arg-szekvenciát tartalmazó peptid alkalmazásával kettős aktivitású gyógyszerek állíthatók elő (2. ábra).

2. ábra. Az Asn-Gly-Arg- (NGR) szekvenciát tartalmazó peptidek kettős receptor-felismerése
Így mind a CD13, mind az integrin-receptorok támadhatók egy vegyület alkalmazásával. Ennek a koncepciónak mentén kezdtük el kísérleteinket ezen a területen.
Az irodalomban két olyan ciklusos peptidet alkalmaztak irányító molekulaként jó eredménnyel, amelyek ezt az AsnGly-Arg tripeptidszekvenciát tartalmazták (3a. ábra). Az egyik egy diszulfidhidat tartalmazó variáns, ahol az Nés C-terminálisra beépített ciszteinek tiolcsoportjait kapcsolták össze diszulfiddá (-CH2-S-S-CH2-) (ciklo[Cys-Asn-Gly-Arg-Cys]), míg a másik variánsban a peptid N-terminálisára beépített lizin (Lys) a-aminocsoportja és a C-terminálisra beépített glutaminsav (Glu) oldalláncának karboxilcsoportja között alakítottak ki amidkötést (-NHCO-) (ciklo[Lys-Asn-Gly-Arg-Glu]-NH2). Mindkét variáns 17 atomot tartalmazott a ciklusban. Korábbi kísérleteink azt mutatták, hogy ha a ciklust tioéterkötéssel (-CH2-S-CH2-) alakítjuk ki, akkor a ciklopeptidek sokkal stabilabbak a kémiai és biológiai lebontásokkal szemben. Ráadásul a tioéterkötés kialakítása kemoszelektív eljárás, ami könnyen, jó kitermeléssel megvalósítható például egy aminocsoporthoz kapcsolt klóracetilcsoport és egy cisztein tiolcsoportja között. Munkánk során öt új tioéterkötést tartalmazó ciklikus peptidet készítettünk el, amelyek felépítése a 3b. ábrán láthatók. Kémiai okok miatt, az irodalmi variánsoknak megfelelő, a ciklusban 17 atomot tartalmazó ciklopeptidet még nem sikerült előállítani.
Vizsgáltuk az előállított új vegyületek aszparaginil-glicin (Asn-Gly) részletének átalakulását aszpartil-glicin(Asp-Gly-), illetve izoaszpartil-glicin(izoAsp-Gly-) egységet tartalmazó származékokká különböző kémiai reakciókhoz használt pufferoldatokban és a sejtes vizsgálatokhoz használt körülmények között. A kapott eredményeket öszszehasonlítottuk a vizsgálatokba bevont két irodalmi analóggal is. A stabilitási vizsgálatokat fordított fázisú, nagyhatékonyságú folyadék-kromatográfia (RP-HPLC) segítségével végeztük. A ciklopeptidek stabilitása jelentősen eltért egymástól. Általánosan megállapítható volt, hogy az irodalmi adatoknak megfelelően pufferoldatban a vegyületek könnyebben átalakultak, mint tiszta vizes oldatban és a pH emelése (az oldat lúgosítása) szintén fokozta a bomlást. A leggyorsabb átalakulást a biológiai vizsgálatok körülményei között tapasztaltuk, feltehetőleg elsősorban a magasabb hőmérsékletnek (37 °C) köszönhetően. Ami viszont elsőre meglepőnek tűnt az, hogy ciklopeptidek szerkezete jelentősen befolyásolta az átalakulás sebességét.
A két irodalmi vegyület a legtöbb vizsgálati körülmény között jelentős stabilitást mutatott. A 2-es számú vegyület (ciklo[Lys-Asn-Gly-Arg-Glu]-NH2) kiugróan nagy stabilitással rendelkezett, és még a biológiai vizsgálatok körülményei között is csak 24 óra elteltével volt jelentősebb bomlás megfigyelhető. Az általunk előállított tioéterkötést tartalmazó analógok többsége lényegesen nagyobb százalékban alakult át aszpartilés izoaszpartil-származékká. Különösen labilisnak mutatkozott a 3-as vegyület (ciklo[CH2-CO-Asn-Gly-Arg-Cys]NH2), amely már savas pH-n is bomlott, és a 7-es vegyület (ciklo[CH2-CO-Lys-Asn-Gly-Arg-Cys]-NH2), amely semleges és lúgos pH-n mutatott szignifikáns átalakulást. Mivel a két ciklopeptid közül az egyik 15, míg a másik 18 atomot tartalmazott a gyűrűben, ez jelezte, hogy nem magának a ciklus méretének, feszültségének van szerepe az átalakulás sebességében. Különösen érdekes volt az a megfigyelés, hogy a két, a gyűrűben 16 atomot tartalmazó vegyület stabilitásában jelentős a különbség. Pedig csak a kénatom helyzete változott meg a gyűrűben (3b. ábra). A 4-es vegyület sokkal stabilabbnak bizonyult, mint a homociszteint tartalmazó analóg (5-ös vegyület).


3. ábra. Az Asn-Gly-Arg tripeptid-szekvenciát tartalmazó ciklopeptidek sematikus szerkezete és az alkalmazott jelölések
Feltételeztük, hogy az észlelt stabilitásbeli különbségek mögött térszerkezeti hatások lehetnek. Ezért először elektronikus cirkuláris dikroizmus (ECD) spektroszkópiával vizsgáltuk az NGR ciklopeptidek szerkezetét. (A méréseket az ELTE Kiroptikai Szerkezetvizsgáló Laboratóriumában Majer Zsuzsa és Knapp Krisztina végezte Jasco J-810 spektropolariméteren). Bár ezek a vizsgálatok nem adtak egyértelmű képet a vegyületek konformációjáról, azt már ezekből a vizsgálatokból is megállapíthattuk, hogy a ciklopeptidek térszerkezete jelentősen eltér egymástól. A 2-es számú vegyület (ciklo[Lys-Asn-Gly-Arg-Glu]-NH2) spektruma különösen eltért a többi vegyület spektrumától. Továbbá megállapítható volt, hogy az átalakulásra kevésbé hajlamos ciklopeptidek szerkezete merevebb, míg a könnyen bomló származékok flexibilis szerkezettel rendelkeznek.

4. ábra. Az Asn-Gly-Arg tripeptid-szekvenciát tartalmazó ciklopeptidek bomlékonysága és térszerkezete közötti összefüggés
A pontosabb térszerkezet felderítése érdekében mágneses magrezonancia (NMR) spektroszkópiai vizsgálatokat is végeztünk. (A méréseket Perczel András professzor irányításával az MTA–ELTE Fehérjemodellező Kutatócsoportban Láng András és Czajlik András végezte Bruker Avance III 700 MHz-es spektrométeren). A ciklopeptidek pontos konformációjának meghatározása két kulcsfontosságú megállapításra vezetett a vegyületek stabilitása és szerkezete közötti összefüggésben. Az első az volt, hogy az aszparagin oldalláncának karbonil (CO) szénatomjának és a glicin nitrogénjének távolsága döntően befolyásolja a szukcinimid-gyűrű kialakulásának sebességét, ezáltal az átalakulási folyamatot. Ennek megfelelően az ciklopeptidek átalakulása aszpartil és izoaszpartil származékokká azokban a vegyületekben a legszámottevőbb, ahol ez a távolság kicsi, míg a távolság növekedése a stabilitást fokozza (4. ábra). Az említett két atom távolsága nem függ a ciklust alkotó atomok számától. A két ciklopeptid, amelyekben a legközelebb helyezkedik el ez a két atom, a gyűrűben 15 atomot tartalmazó ciklo[CH2CO-Asn-Gly-Arg-Cys]-NH2 és a 18 atomot tartalmazó ciklo[CH2-CO-Lys-AsnGly-Arg-Cys]-NH2 (2,84 A, illetve 2,87 A). Ráadásul pont ez a két vegyület mutatkozott az ECD vizsgálatokban is a legmozgékonyabbnak, így a szukcinimid-gyűrű kialakulásának nincs szerkezeti gátja. Ezt támasztotta alá az is, hogy ezen vegyületek esetében az NMR-szerkezetek nem mutattak a konformáció stabilizálását biztosító hidrogénkötést a peptidgerinc atomjai között. Ahogy az említett két atom távolsága nő a ciklopeptidekben, úgy nő a vegyületek stabilitása is (4. ábra), amelyet tovább fokozhat a molekulán belül kialakuló hidrogénkötés is. A sorból itt is kilóg a 2-es vegyület, amelyben a két adott atom távolsága csak 3,79 A. A korábbi megállapításaink alapján azt lehetett volna várni, hogy ennek vegyületnek közepes lesz a stabilitása, hasonlóan a homociszteint tartalmazó tioéteres variánshoz (5-ös vegyület). Azonban a vizsgálatok a vegyület extrém stabilitását mutatták. A magyarázat erre, hogy mind az aszparagin oldalláncának karbonilcsoportja, mind a glicin NH-csoportja erős hidrogénhidas szerkezetben stabilizálódik, így igen csekély az esély arra, hogy – a köztük lévő nem jelentős távolság ellenére – ezek a csoportok tovább közelítsenek egymáshoz. Mindezek alapján azt mondhatjuk, ez a ciklopeptid a legalkalmasabb arra, hogy szelektíven csak a CD13-receptoron keresztül juttassunk hatóanyagot a tumorsejtekbe vagy tumordiagnosztikában (PET: pozitron emissziós tomográfia) alkalmazzuk. Ugyanakkor a kevésbé stabil vegyületek alkalmasak lehetnek az általunk célként kitűzött „egy anyag két célpont” alapú irányított daganatterápiára. Ezért előkísérleteinkben a két legbomlékonyabb, tioéterkötést tartalmazó 3-as és 7-es vegyületet (ciklo[CH2-CO-Asn-GlyArg-Cys]-NH2 és ciklo[CH2-CO-Lys-Asn-Gly-Arg-Cys]-NH2), valamint a diszulfidhidat tartalmazó stabilabb ciklopeptidet (Acciklo[Cys-Asn-Gly-Arg-Cys]-NH2) választottuk a hatóanyagot tartalmazó konjugátumok előállítására.
A konjugátumokat (a daganatellenes szer irányító peptidhez kapcsolt formája) úgy alakítottuk ki, hogy a ciklopeptidet két glicin aminosavon, mint távolságtartón keresztül egy lizin e-aminocsoportjához kapcsoltuk. A lizin a-aminocsoportjához pedig egy enzimlabilis tetrapeptid spaceren keresztül (Gly-Phe-Leu-Gly) aminooxiecetsavat (Aoa) kötöttünk, amelyhez oxim ligációval kapcsoltuk a daunomicint, ami egy gyakran alkalmazott kemoterápiás szer (5. ábra).

5. ábra. A daunomicint és irányító peptidet tartalmazó konjugátum sematikus szerkezete
Az enzimlabilis távtartó (spacer) egy a tumorsejtekben túltermelődő enzim (Katepszin B) hatására hasad és egy aktív metabolit keletkezik, ami vizsgálataink szerint kötődik a DNS-hez, ezáltal gátolja a tumornövekedést. A konjugátumok hatékonyságát két ráksejten vizsgáltuk Szakács Gergely és Tóth Szilárd (MTA–TTK) segítségével. Az egyik tartalmazott CD13-receptort (HT-1080 fibroszarkóma), a másik nem, de integrin-receptorokat igen (HT-29 vastagbél adenokarcinóma). Az előzetes kísérletek azt mutatják, hogy a legjobb hatást az a konjugátum mutatta, amelyben a daunomicint a ciklo[CH2-CO-Lys-Asn-Gly-Arg-Cys]-NH2 irányító peptidhez kapcsoltuk. A daganatsejtek szaporodásának gátlása némileg nagyobb volt a fibroszarkóma sejteken (CD13 pozitív sejtek), mint a vastagbél tumorsejteken (CD13 negatív, integrin receptor pozitív), de nincs jelentős különbség a mutatott hatásban. Ez arra utal, hogy a konjugátum mindkét receptoron kifejtheti hatását. Annak érdekében, hogy a konjugátumnak az adott receptorokhoz való kötődését és ezeken keresztül történő sejtbe jutását igazoljuk, a továbbiakban a receptorok blokkolására alkalmas ciklopeptideket fogunk alkalmazni gátlási kísérletekben. Reményeink szerint ezzel igazolni tudjuk a kiválasztott konjugátum(ok) két receptoron történő hatását, amely a molekula időben történő átalakulásának következménye. Amennyiben az in vitro vizsgálatok sikerrel zárulnak, a vegyületek hatékonyságát tumoros állatmo-delleken is igazolni kívánjuk.
Az OTKA (K 104045) pályázat keretében végzett munkánk és elért eredményeink alapján meghívást kaptunk egy konzorciális Horizon 2020 EU-s pályázatba is (MARIE SKŁODOWSKA-CURIE ACTIONS Innovative Training Networks), amelynek fantázianeve MAGICBULLET (mágikus golyó). A 2015 januárjában indult és 4 év időtartamú pályázatban az Eötvös Loránd Tudományegyetem mellett német (Bielefeld, Köln), olasz (Milánó, Como) és finn (Helsinki) egyetemek és intézetek (Országos Onkológiai Intézet és a Kineto Lab Magyarországról, a Heidelberg Pharma GmbH, a Bayer Pharma AG és Optical Imaging Centre Erlangen Németországból, valamint az Exiris és a Nerviano Medical Science Olaszországból) vesznek részt. A konzorcium tagjai a tématerület jelentős művelői és reményeink szerint a kutatócsoportoknak ez a nemzetközi együttműködése nemcsak a tudás, hanem az előállított vegyületek szinergiáját is magával hozza, magas szintű tudományos közleményeket eredményezve. A konzorcium magyar tagjainak munkáját Mező Gábor (MTA-ELTE) és Tóvári József (OOI) fogja irányítani.
A cikkben használt egyés hárombetűs aminosavkódok és további rövidítések jelentése: Aszparagin: N (Asn); aszparaginsav: D (Asp); arginin: R (Arg); cisztein: C (Cys); glicin: G (Gly); leucin: L (Leu); lizin: K (Lys); fenilalanin: F (Phe); aminooxi-ecetsav (Aoa); prolin; P (Pro), homocisztein, hC(homoCys); daunomicin (Dau).
Irodalom
1. Mező, G. (2011) Célzott
tumorterápia peptidekkel. Természet Világa 142, 555-558.
2. Mező, G., Hegedüs, R.
Szabó, I. (2012) Célzott tumorterápia. Természet Világa 142, 448-451.
3. Healy, J. M. és mtsai.
(1995) Peptide ligands for integrin alpha v beta 3 selected from random
phage display libraries. Biochemistry 34, 3948–3955.
4. Curnis, F. és mtsai.
(2002) Differential binding of drugs containing the NGR motif to CD13 isoforms
in tumor vessels, epithelia and myeloid cells. Cancer Res. 62, 867–874.
5. Corti, A.; Curnis, F.
(2011) Tumor vasculature targeting through NGRpeptide-based drug delivery
systems. Curr. Pharm. Biotechnol. 12, 1128–1134.
6. Geiger, T., Clarke, S.
(1987) Deamidation, isomerisation, and racemisation at asparaginyl and
aspartyl residues in peptides. J. Biol. Chem. 262, 785–794.
7. Colombo, G. és mtsai.
(2002) Structureactivity relationship of linear and cyclic peptides containing
NGR tumor-homing motif. J. Biol. Chem. 277, 47891–47897.
8. Negussie, A. H. és mtsai.
(2010) Synthesis and in vitro evaluation of cyclic NGR-peptide targeted
thermally sensitive liposome. J. Control. Release 143, 265–273.
9. Enyedi, K. N. és mtsai.
(2015) Development of cyclic NGR peptides with thioether linkage: structure
and dynamics determining deamidation and bioactivity. J. Med. Chem. 58,
1086-1817.
10. Orbán, E. és mtsai.
(2011) In vitro degradation and antitumor activity of oxime bond-linked
daunorubicin-GnRHIII bioconjugates and DNA-binding properties of daunorubicin-amino
acid metabolites. Amino Acids 41, 469-483.
11. Máté, G. és mtsai (2015)
In vivo imaging of Aminopeptidase N (CD13) receptors in experimental renal
tumors using the novel radiotracer 68Ga-NOTA-c(NGR). Eur. J. Pharm. Sci.
69, 61-71.
| Természet Világa, | 146. évfolyam, 7. szám,
2015. július
http//www.termvil.hu/ |