|
Tumorevolúció és terápiás lehetőségek
A
szervezetünket felépítő sejtek a legtöbb esetben tudatos közreműködésünk
nélkül látják el feladataikat. Ha megvágjuk magunkat, a sejtjeink osztódni
kezdenek, differenciálódnak, majd kis idő múlva a seb összezáródik és beforr.
Ha fertőzés ér bennünket, a sejtjeink ellenanyagokat termelnek, és megpróbálják
ártalmatlanná tenni a betolakodókat.
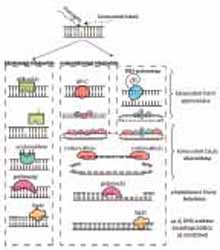
Molekuláris szinten elsősorban a fehérjék végzik a sejtek és a szervezet működéséhez szükséges – a fenti példákhoz hasonló – feladatokat. Azt az információt, hogy fehérjéink pontosan hogyan épüljenek fel, egy hosszú, négyféle nukleotidból álló molekula, a DNS tárolja. Ezek alapján azt hihetnénk, hogy a DNS igen stabil molekula, és a benne tárolt információ állandó, de ez koránt sincs így. Egy átlagos napon minden sejtünkben a DNS-t felépítő nukleotidból több tízezer módosul úgy, hogy a módosulás megváltoztathatja a tárolt információt, és annak értelmét. Ezek egy részét külső behatás okozza, ilyen az UV-sugárzás, amely a DNS-ben lévő egymással szomszédos timin vagy citozin bázisok között hozhat létre kovalens kötést, vagy akár a dohányfüst egy komponense, amely a guanin bázisokra képes egy metilcsoportot építeni. A változások másik forrása azonban belső eredetű, a sejt normális anyagcseréje során elkerülhetetlenül keletkező reaktív ágensek okozzák. Ezek a módosulások két esetben jelentenek komoly veszélyt a sejtre. Az első, ha az adott DNS-szakaszról RNS-átírás (transzkripció) történik. Az átírást egy RNS-polimeráz nevű enzim végzi. Ez végighalad a DNS-szálon, és azt mintaként felhasználva, létrehozza azt az RNS-molekulát, ami nélkülözhetetlen a sejt számára megfelelő fehérje szintéziséhez. Ha a DNS-en rendellenesség adódik, az RNS-polimeráz nem mindig lesz képes a helyes információt kódoló RNS átírására, így arról nem fog a sejt számára funkcióképes fehérje képződni. A másik eset, amikor a sejt osztódik, és megkettőződik a DNS-e. Ilyenkor egy úgynevezett DNS-polimeráz enzim halad végig a DNS szálain. Ennek feladata a nagy pontosságú másolás. Ha módosult bázishoz ér, azt nem mindig képes felismerni, és a másolás megakadhat. Ilyen esetben az adott helyen a DNS-szál el is törhet, vagy a másolása nem fejeződik be a rendelkezésre álló idő alatt, és a sejt elpusztulhat, vagy genetikai változásokat szenvedhet. DNS-hibajavító folyamatok Természetesen a sejt védekezik a módosulások ellen, méghozzá úgy, hogy folyamatosan ellenőrzi a DNS-t, és a megváltozott nukleotidokat eltávolítja, helyükre pedig sérülésmentes nukleotidokat épít be. Az ezért felelős rendszereket összefoglaló néven DNS-hibajavító mechanizmusoknak nevezzük. A DNS-hibajavítás jelentőségét az is jól mutatja, hogy jelenleg már több mint 300 különböző fehérjének tulajdonítunk szerepet a DNS-ben tárolt információ integritásának megőrzésében. A DNS módosulásainak típusa – az említett néhány példán túl – szinte végtelen lehet. A sejtben velük foglalkozó rendszereket legtöbbször a hibafelismerés módozata alapján osztjuk csoportokba. A gyakran előforduló módosulásokra léteznek olyan fehérjék, amelyek közvetlenül felismerik azokat. Ezt a csoportot báziskivágó hibajavításnak nevezzük (1. ábra). A felismerés után a módosult bázist leválasztják a DNS cukor-foszfát gerincéről, majd bemetszést generálva, kivágják az immár bázis nélküli (ún. abázikus) nukleotidot. A következő lépésben egy, a DNS másolását végző polimerázhoz nagyon hasonló enzim kitölti a folytonossági hiányt a megfelelő nukleotiddal, a másik ép DNS-mintaszálat másolva, így visszaáll a módosulás előtti információtartalom.1 Tekintettel a DNS-hibák sokféleségére, a sejtnek nem gazdaságos minden egyes hibára fenntartani egy olyan fehérjét, amely azt speciálisan felismeri. Az úgynevezett nukleotidkivágó hibajavító mechanizmusok sokkal általánosabban működnek (1. ábra). Ahelyett, hogy a felismerő fehérjék egy-egy speciális hibát keresnének, sokkal inkább a DNS általános alakját vizsgálják, miközben teljes hosszában folyamatosan pásztázzák a DNS-t, és azokat a részeket azonosítják, amelyek eltérnek a normálistól. Működésükre jellemző, hogy nemcsak a károsodott nukleotidot távolítják el, hanem egy DNS-szakaszt is a hiba környezetében. Ezután egy DNS-polimeráz tölti fel a hiányzó részt (1. ábra).2
1. ábra. Néhány DNS-hibajavító útvonal Még ennél is energiatakarékosabb megoldás az, amikor a sejt a hibánál elakadt RNS-polimerázt azonosítja, hiszen így még a módosult szerkezet keresésébe sem kell energiát fektetnie, ráadásul az éppen átíródó, vagyis az adott pillanatban legfontosabb terület meghibásodását javíthatja. Adódhatnak olyan szituációk, amikor olyan nagy a DNS-károsodás mértéke, hogy az említett rendszerek nem képesek az összes hiba eltávolításra. Így a módosulások megmaradnak addig, amíg a DNS megkettőződésére kerül a sor. Mivel a folyamatot végző DNS-polimeráz csak az ép nukleotidokon képes áthaladni, hiba esetén a másolás elakadhat. Ilyenkor az úgynevezett hibatolerancia mechanizmusok lépnek működésbe. Ezek feladata, hogy átjuttassák a másolást végző replikációs komplexet az akadályt jelentő módosuláson. Ekkor a nagy pontosságú DNS-polimeráz szerepét olyan ún. DNS-hibaátíró polimerázok veszik át, amelyek képesek nukleotidot illeszteni a hibás bázisokkal szemben is. Bár a módosulás így megmarad, ezt egyéb javító mechanizmusok a későbbiekben még eltávolíthatják. A sejt számára a problémát inkább az jelenti, hogy sok esetben a módosult bázissal szemben már nem az eredeti információtartalmat jelentő nukleotid épül be, és az információtartalom visszafordíthatatlanul és végérvényesen megváltozik. Ezt nevezzük mutációnak. A mutáció azon kívül, hogy az evolúció hajtóereje, közvetlenül felelős a karcinogenezisért, vagyis a sejtek rákos elváltozásáért. DNS-hibajavításban szerepet játszó génekhez köthető betegségek A különféle DNS-hibajavító mechanizmusok működése elengedhetetlen a sejt életben maradásához. Ezekben a folyamatokban számos fehérje, fehérjekomplex vesz rész, bonyolult hálózatokat alkotva. Ezek a fehérjék ugyanúgy keletkeznek, mint a sejtjeink összes egyéb folyamataiban résztvevő fehérjéi, így jogosan merülhet fel bennünk a kérdés, hogy mi történik, ha az őket kódoló DNS-szakaszban következik be valamilyen változás. Ha a javítást végző fehérjék, fehérjekomplexek egyes tagjai hiányoznak, vagy hibásan működnek, súlyos betegségek léphetnek fel, amelyek közös jellemzője lehet például a rákra való hajlam, a fényérzékenység, és/vagy a korai öregedés. A betegek egy részénél előfordulhat egyéb idegrendszert (látás, hallás, mozgás) érintő rendellenesség is.3 A nukleotidkivágó hibajavító mechanizmusokban résztvevő fehérjekomplex valamely tagjának hiánya a Xeroderma pigmentosum (XP) betegség kialakulásához vezethet. A betegségben szenvedők különösen érzékenyek a napsugárzásra. Számukra akár pár perces napfény is hetekig tartó hólyagos bőrpírt okozhat, míg más esetekben a bőr szárazságát (xeroderma), illetve elszíneződését (pigmentosum) okozza. Esélyük a daganatos megbetegedésre igen nagy (2. ábra)4.
2. ábra. A Xeroderma pigmentosumban szenvedő betegek csak speciális öltözékben tartózkodhatnak a napon, amely megvédi őket a káros sugárzásoktól Az úgynevezett Cockayne-szindróma (CS) hátterében a CSA- és CSB-fehérjék valamelyikének a hiánya áll. Ezek ismerik fel az RNS-polimerázt, mely egy adott hibának köszönhetően nem képes folytatni az átírást. Fehérjéket toboroznak a károsodás helyére, melyek kijavítják a hibát. Ha ez nem történik meg, az RNS szintézise és ebből következőleg a fehérjeszintézis is gátakba ütközik. A betegségre jellemző az idegrendszer, a belső szervek, szervrendszerek, csont, valamint az ízületek nem megfelelő fejlődése, halláscsökkenés, szemrendellenességek és korai öregedés. A sejtosztódásban szerepet játszó BLM-fehérje hiányára vezethető vissza a Bloom-szindróma, melyre növekedési problémák, fényérzékenység, daganatos megbetegedésre való hajlam, illetve csökkent immuntevékenység jellemző. A BLM-fehérje DNS-helikáz aktivitással rendelkezik, mely a DNS-spirál „széttekerését” végzi a hibajavítás során. Ezen kívül a BLM-fehérje kapcsolatba lép más fehérjékkel, melyekkel a DNS stabilitásának fenntartásában vesz részt. Hiányában ezek a mechanizmusok nem működnek, ami nagymértékű kromoszóma-átrendeződésekhez és végül a Bloom-szindróma kialakulásához vezethet. Egy másik DNS-helikáz, a WRN-fehérje abnormális formája okozza a Werner-szindrómát, mely korai öregedést okoz. Ez a genetikai betegség nem túl gyakori, nagyjából tízmillió születésre jut egy eset (3. ábra).
3. ábra. Werner-szindrómás betegeknél a korai öregedés jelei már a késői kamaszkorban jelentkeznek (Forrás: University of Washington) A Rothmund–Thomson-szindrómát is egy meghibásodott DNS-helikáz okozza. A betegség során emésztőrendszeri, csontozat-fejlődési rendellenességek, bőr-, haj-, köröm-, fogkialakulási problémák jelentkezhetnek. Kiemelkedően magas a daganatok kialakulásának kockázata is, különösen csont-, és bőrrák fejlődhet ki. A rák kialakulása Ha a sejt védelmi mechanizmusa a DNS-károsodásokkal szemben zavart szenved, ennek kétféle fő következménye lehet, amelyek az említett betegségeknél is megfigyelhetők. Egyrészt hatással lehet a transzkripcióra, ezáltal a fehérjeszintézist érinti. Ez a szervek, szövetek fejlődésére gyakorol hatás. Másrészt a genetikai állomány gyors változása rák kialakulásához is vezethet. Mit is jelent valójában a rák? A rák egy olyan sejtcsoport, ami kiesik a szervezet ellenőrzése alól, a szabályzó ingerekre nem reagál, és kontrollálatlan osztódásba kezd, ezáltal daganatot képez. Míg a jóindulatú daganat nem képez áttétet, és csak akkor károsítja az életfunkciókat, ha a mérete miatt zavarja a többi szerv működését, addig a rosszindulatú daganat a véráram segítségével eljuthat a szervezet bármely pontjára, és áttéteket képezhet. Miben más a ráksejt, mint az összes többi? Valójában genetikai másságról van szó, ugyanis legtöbbször számos gén sérül, köztük olyanok is, amelyek különös fontosságúak a rák kialakulása szempontjából. Ezek a proto-onkogének és tumorszupresszorok. A tumorszupresszorok funkciója, ahogyan a nevük is erre utal, a tumorképződéssel járó folyamatok vis.szaszorítása. Szerepük normális esetben például a sejtosztódás féken tartása, vagy annak biztosítása, hogy a sejt megfelelő időben a megfelelő módon pusztuljon el. Ha ezek szabályzása zavart szenved, igen gyakran alakul ki rák. A p53 fehérje például tipikus tumorszupresszor. Fehérjeterméke DNS-károsodás hatására leállítja a sejtciklust, időt hagyva a javító mechanizmusoknak a hiba kiküszöbölésére. A p53 a daganatok közel 50%-ában mutációt szenvedett vagy hiányzik. Ha a p53 nem képes betölteni funkcióját, a javító mechanizmusoknak nem lesz elég idejük elvégezni feladatukat, melynek következtében további mutációk halmozódnak fel a sejtben.5
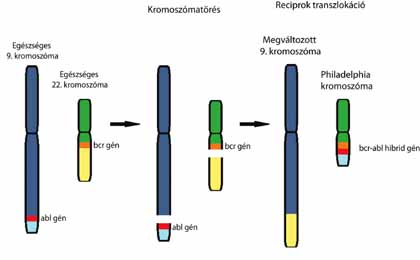
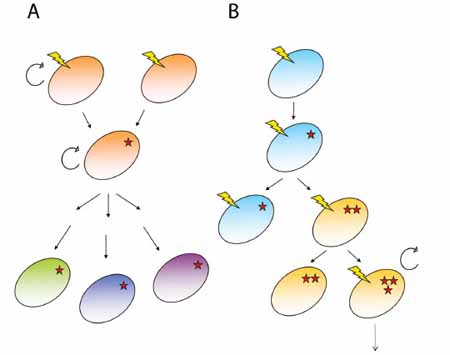
4. ábra. A kromoszómaátrendeződés következménye a bcr-abl hibrid gén, melynek terméke foszforilálció segítségével aktiválni képes egyéb, a sejtosztódás serkentésében szerepet játszó fehérjéket A proto-onkogének az egészséges sejtek normális működéséhez szükségesek, például a sejt osztódását segítik. Ha egy proto-onkogénben mutáció következik be, onkogénné alakulhat. A krónikus mieloid leukémia egyik típusában kimutatták az úgynevezett Philadelphia-kromoszómát. Ebben az esetben a 9. és 22. kromoszómák végei cserélődnek ki egymással (reciprok transzlokáció), melynek következtében egy kisebb méretű 22. (Philadelphia) kromoszóma keletkezik. A csere következtében a kromoszómavégeken elhelyezkedő bcr és az abl gének összeolvadásával egy bcr-abl hibrid gén keletkezik, amely onkogénként viselkedik. A róla képződő fehérje olyan egyéb fehérjék aktiválásában vesz részt, amelyek elősegítik a kontrollálatlan sejtosztódást (4. ábra)6. Tumorevolúció A tumorok kialakulása korántsem egyszerű folyamat. A leírtak alapján megállapíthatjuk, hogy számos ponton történhet hiba a sejtben, ami rákos elfajuláshoz vezethet, de általában egyetlen gén mutációja nem elég ehhez. A sejt több ponton is ellenőzi önmagát, és az immunrendszerünk is monitorozza a sejtjeinket a szervezetünk védelmében. A rendszer azonban nem tökéletes. Esetenként túlél egy-egy sejt, ami daganatot képezhet. A legtöbb tumor vis.szavezethető egyetlen sejtre. Hiba lenne azonban azt hinni, hogy a daganatot alkotó sejtek nem változnak. Egyetlen tumort is általában heterogén ráksejtek populációja alkotja. A különböző sejtösszetételű daganat könnyen alkalmazkodik a környezeti változásokhoz, tartalmazhat olyan sejteket, melyek ellenállóak az alkalmazott terápiákra, ami a betegség kiújulásához vezethet. A folyamat hátterét két elmélet magyarázza: a klonális evolúció és a tumorőssejt-modell. A tumorőssejt-hipotézis állítása szerint a daganat egyes sejtjei őssejthez hasonló tulajdonságúak, így irányítják a tumor kialakulását, fejlődését, terjedését vagy kiújulását. Ezeknek a sejteknek – egy egészséges őssejthez hasonlóan – korlátlan önmegújító képességük van, és differenciálódásra is alkalmasak. Ezek következtében a tumorőssejtek képesek különböző tulajdonságú sejteket létrehozni, így kialakítva a heterogén tumort. Ezen modell alapján az áttétek kialakulásáért a tumorőssejtek terjedése tehető felelőssé, míg a rák kiújulása a tumorőssejt-terápiára mutatott rezisztenciájának eredménye (5A. ábra).
5. ábra. A tumorok keletkezését és fejlődését magyarázó elméletek modelljei. A tumorőssejt-modell (A) és a klonális evolúció hipotézise (B). A villám a DNS-károsítást, a csillag a létrejött mutációt jelzi (az első csillag minden esetben azt a többszörös mutációt jelenti, amely következtében létrejön egy ráksejt) A klonális evolúció elmélete alapján a daganat sejtjei idővel mutációk különböző kombinációira tesznek szert, így lépésről lépésre kiválasztódnak a legmegfelelőbb, legagresszívabb sejtek, amelyek irányítják a tumor fejlődését. Eszerint a tumor kialakulása egy adott sejtben jelentkező többszörös mutáció megjelenésével történik. A tumor fejlődése során a genetikai instabilitás és a kontrollálatlan osztódás miatt létrejöhetnek újabb mutációkat hordozó sejtek, újabb tulajdonságokkal. Ezek a sejtek további nagy mennyiségű, azonos tulajdonságokkal rendelkező utódsejtet hozhatnak létre, vagy további mutációkat szerezhetnek, növelve a tumor heterogenitását. Az újonnan keletkező, újabb mutációkon átesett sejtek még nagyobb növekedési előnyre tesznek szert, ezúttal más tumorsejtekkel szemben. A klonális evolúció hipotézise alapján számos ráksejttípusból kialakulhat olyan sejt, amely potenciálisan áttétet képezhet, ellenállóvá válhat a terápiákra és hozzájárulhat a betegség kiújulásához (5B. ábra).
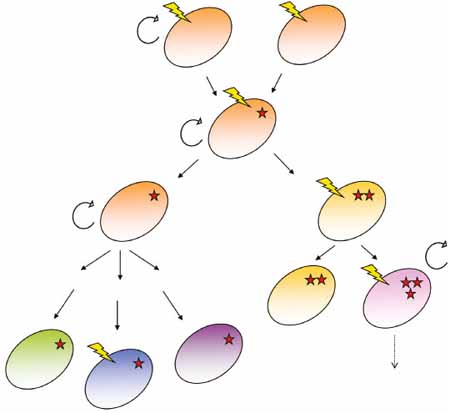
6. ábra. A tumorőssejt-modell és a klonális evolúció hipotézise szerint a leírt folyamatok – napjaink mellrákra irányuló kutatásai alapján – a tumor heterogenitásáért egyaránt felelősek7 A tumorőssejt-modell és a
klonális evolúciós hipotézis is egyetért abban, hogy a tumor egyetlen,
többszörös mutáción átesett sejtből ered, mely később korlátlan osztódásra
és utódképzésre válik képessé. A két hipotézis jól megfér egymás mellett.
A mellrák kutatása során végzett tanulmányok alapján a tumorok heterogenitásáért
felelős folyamatok a klonális evolúció mellett a tumorőssejt-modell jellegzetességeit
is mutatják. Eszerint a differenciáció és a klonális szelekció kombinációja
vezet az önmegújító képességgel rendelkező sejtek kialakulásához. A tumort
alkotó különböző sejtek között lehetnek differenciáltak, melyek szaporodó
képessége csökkent, és olyanok is, melyek fokozott önmegújító és szaporodó
képességgel rendelkeznek (6. ábra).
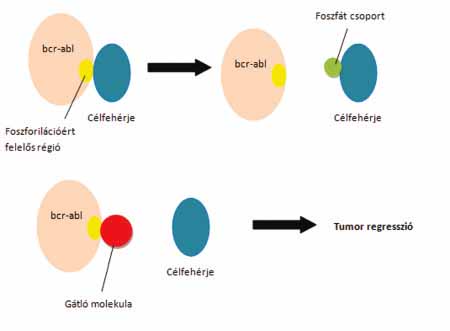
A daganatokkal szembeni harcot megnehezíti az a tény, hogy a ráksejtek a legtöbb tulajdonságukban megegyeznek a testi sejtjeinkkel, hiszen azokból alakulnak ki. Létezik azonban néhány olyan tulajdonság, melyekben eltérnek, ilyen a gyors osztódási képesség vagy a megnövekedett energiaigény. A hagyományos gyógyszeres terápiák ezeket a lehetőségeket próbálják kiaknázni. A kemoterápiáról a legtöbb embernek a daganatos megbetegedések kezelése, a ráksejtek elpusztítása jut eszébe. Ez a fogalom azonban nemcsak erre vonatkozik, hanem összefoglaló neve minden betegség gyógyszeres kezelésének. A műtéttől és a sugárkezeléstől abban tér el, hogy majdnem mindig szisztematikus eljárásként használják. Míg a sugárkezelés a testnek csak egy bizonyos részén fejti ki hatását, addig a kemoterápia esetében a gyógyszernek általában az egész testen át kell haladnia, mire eljut a ráksejtekhez. A terápia során az első és legfontosabb cél a ráksejtek elpusztítása, és visszatérésének megakadályozása. Azonban előfordulhat, hogy a betegség már olyan stádiumban van, ahol ez nem lehetséges. Ilyenkor a kontrollálás válik fontossá, tehát a terjedés és növekedés megakadályozása, illetve a daganat méretének csökkentése. Előrehaladott állapotban a tünetek enyhítése, és az életminőség javítása kap központi szerepet. A kemoterápia alkalmazásának időpontját a betegség stádiuma nagyban befolyásolja. Az orvosok a rák típusa, a beteg egészségi állapota és kora alapján mérik fel, hogy milyen szereket, mekkora dózisban és mennyi ideig használjanak a terápia során. A gyógyszerek bejuttatására több lehetőség van. Történhet szájon át, intravénásan, izomba adva vagy bőr alá injekciózva. Az intravénás a leggyakrabban használt módszer. Az orális bevitel hátránya, hogy az emésztőrendszer sok esetben nem engedi a felszívódást, míg a többi lehetőség esetében gyakran bőr vagy izom irritáció léphet fel. A kemoterápia során a betegek többségének életét a gyógyszerek okozta mellékhatások is nehezítik. A további nehézségek elkerülése végett fontos, hogy a betegek tájékoztassák kezelőorvosukat minden általuk szedett gyógyszerről, étrend kiegészítőről, vitaminról, ezek ugyanis kölcsönhatásba léphetnek a kemoterápiás szerekkel, és csökkenthetik, illetve módosíthatják hatásukat.8 Az elmúlt években nagy hangsúlyt kapott az úgynevezett célzott terápia. A hagyományos kemoterápia legtöbb esetben nem specifikus módon gátolja a sejtek osztódását, és azt használja ki, hogy a ráksejtek gyorsabb osztódásra képesek egészséges társaikhoz képest. Azonban a szervezetünkben is találhatóak olyan sejtek, amelyeknek nagyobb az osztódási rátájuk, ilyenek például a haj növekedésében szerepet játszó sejtek. Ezek a hagyományos kemoterápia során szintén nagymértékben pusztulnak, és szervezetünk más sejtjei is sérülhetnek. Ezzel ellentétben, a célzott terápia általában olyan molekulák gátlásán keresztül történik, amelyek a tumor fejlődéséhez és növekedéséhez nélkülözhetetlenek (7. ábra). A célzott terápia olyan betegek számára is reményt jelenthet, akiknél más kezelés hatástalan. Ezt a módszert általában a hagyományos kezelésekkel kombinálva, azok mellett, és nem pedig helyette alkalmazzák.9
7. ábra. Inhibitor alkalmazása krónikus mieloid leukémia kezelésére. A bcr-abl foszforilációért felelős régiójához kapcsolódni képes a gátló molekula. Ennek köszönhetően az nem tudja betölteni funkcióját, így megszűnik a jel a korlátlan sejtosztódásra Igazán hatásos módszer az úgynevezett személyre szabott terápia lenne. Ennek alkalmazásakor személyre szabott molekuláris diagnózis felállítása szükséges. Ez azonban igen nagy feladat, ismerve a tényt, hogy az egyes tumorok, sőt a tumorokat alkotó sejtek genetikai háttere is eltérő lehet. A daganatos megbetegedések milliók haláláért felelősek évente. A tudomány előre haladásával egyre többet tudunk meg a hátteréről. Legtöbb esetben a rák kialakulásáért a DNS-ben felhalmozódó hibák felelősek. Az evolúció során kialakultak olyan DNS-hibajavító mechanizmusok, melyek bonyolult fehérjehálózatok segítségével igyekeznek féken tartani a mutációkat, és helyreállítani a genom stabilitását. A folyamat azonban nem mindig tökéletes, és a rizikófaktorok (dohányzás, UV-fény, egészségtelen életmód) mindennapos halmozásával túlterheljük őket, ezért felgyorsul a DNS-hibák és -mutációk keletkezésének a sebessége, amely gyakran vezet az egészséges sejtek rákos transzformációjához és daganatok képződéséhez. A rák elleni küzdelmet tehát nagyban nehezíti, hogy igen heterogén betegségről van szó, ugyanis kimondható, hogy minden daganat egyedi, ráadásul a terápia során valójában a saját módosult sejtjeinket kell elpusztítanunk. Az a tény, hogy ezek a sejtek az idő előrehaladtával is további genetikai változásokon mehetnek keresztül, még inkább bonyolítja a terápiás lehetőségeket. Mindezek miatt fontos, hogy minél több molekuláris részletét feltárjuk a DNS-mutációk kialakulásának, valamint ezek hatását a karcinogenezisre, hiszen a rákkal folytatott küzdelemben csak akkor van esélyünk nagy biztonsággal győzni, ha megismerjük a kialakulásának pontos részleteit. Á DÖME LILI– BERCZELI ORSOLYA– DEMCSÁK ANETT–PINTÉR LAJOS SZUKACSOV VALÉRIA–HARACSKA LAJOS Irodalom [1] Robertson AB, Klungland A, Rognes T, Leiros I.: DNA repair in mammalian cells: Base excision repair: the long and short of it., Cell Mol Life Sci., 2009 [2] Wouter L. de Laat, Nicolaas G.J. Jaspers, Jan H.J. Hoeijmakers: Molecular mechanism of nucleotide excision repair, Genes & Dev., 1999 [3] Lisa Wiesmüller, James M. Ford, Robert H. Schiestl: DNA Damage, Repair, and Diseases, J Biomed Biotechnol., 2002 [4] Lehmann AR, McGibbon D, Stefanini M.: Xeroderma pigmentosum, Orphanet J Rare Dis., 2011 [5] A Sigal, V Rotter: Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome, Cancer Res, 2000 [6] Claus R. Bartram, Annelies de Klein, Anne Hagemeijer et al.: Translocation of c-abl oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia, Nature, 1983 [7] Lauren L. Campbell, Kornelia Polyak: Breat Tumor Heterogenity, Cancer Stem Cells or Clonal Evolution?, Cell Cycle, 2007 [8] American Cancer Society: Chemotherapy Principles, An In-depth Discussion of the Techniques and Its Role in Cancer Treatment, 2013 [9] Gerber DE.: Targeted
therapies: a new generation of cancer treatments, Am Fam Physician, 2008
A kutatásokat az OTKA 101225
számú pályázata támogatja.
|