Az érzőideg-végződésekből aktiváció hatására jelátviteli anyagok, köztük számos fehérje természetű, peptid szerkezetű anyag felszabadul, amelyek az erekre és az immunsejtekre hatnak. Ennek eredményeképpen az aktivált immunsejtekből felszabaduló anyagok visszahatnak az érzőideg-végződésekre, azok érzékenységét fokozhatják, vagy éppen gátolhatják. E területek az utóbbi időben a nemzetközi kutatási irányok fókuszába kerültek a hosszan tartó, elsősorban idegi eredetű, neuropátiás fájdalom kezelési nehézségei és új hatásmechanizmusú fájdalomcsillapító gyógyszerek fejlesztési igényei miatt.
Kutatócsoportunk évtizedek óta a krónikus fájdalom komplex mechanizmusait és az érző idegrendszer, immunrendszer és érrendszer közti összetett kapcsolatrendszereket vizsgálja. A traumás (baleset, műtét), metabolikus (cukorbetegség) vagy toxikus (kemoterápia, alkohol, vagy egyéb) eredetű idegsérülés következtében kialakuló neuropátiás fájdalomállapotok nagy betegpopulációt érintenek, ahol a terápia nem megoldott. Ezekben a betegekben a széles körben elérhető, sokféle klasszikus nem-szteroid típusú gyulladáscsökkentő/fájdalomcsillapító gyógyszer nem hatékony, az opioid vegyületek (morfin és származékai) nem vagy csak nagy dózisban fejtenek ki hatást. Az adjuváns analgetikum csoportba sorolható, elsősorban epilepszia és depresszió ellenes gyógyszerek bizonyos betegeknél csökkentik ugyan a fájdalmat, de széles körű, súlyos mellékhatásaik miatt nem lehet őket hosszú távon alkalmazni (Botz és mtsai., 2017). Mindezek alapján tehát a krónikus neuropátiás fájdalom gyógyszerfejlesztési szempontból kiemelt kutatási téma.
Fókuszban a kapszaicin-érzékeny érző idegrendszer
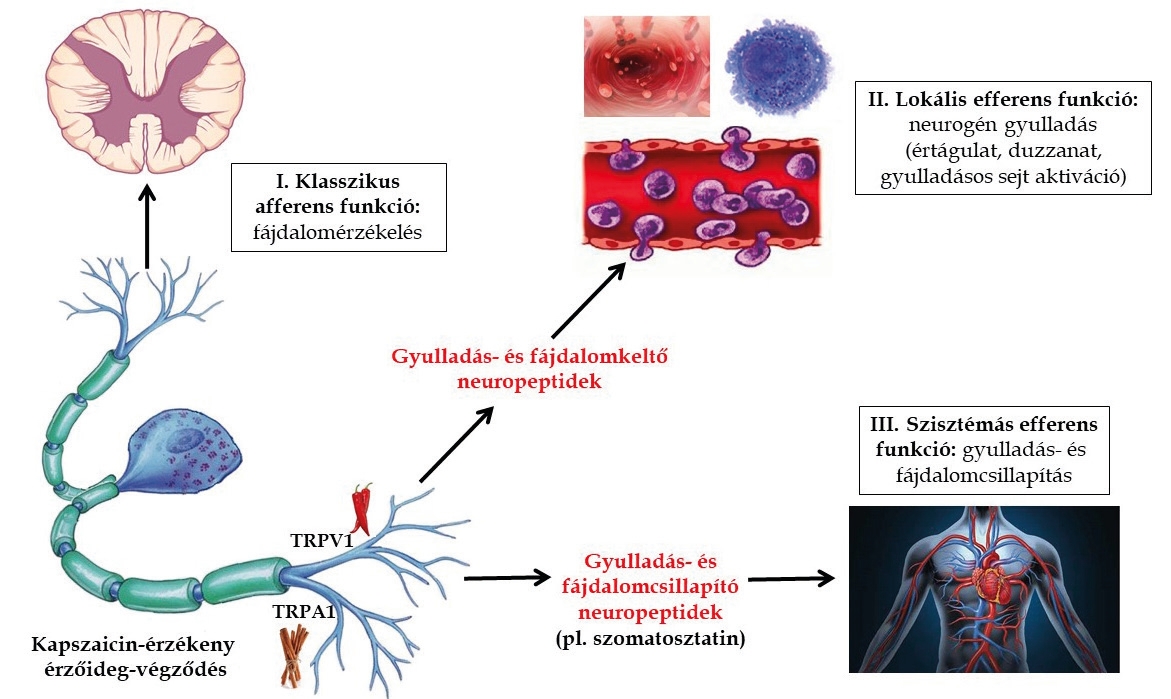
A paprika (Capsicum annuum) nemcsak a magyaros konyha jellegzetes fűszere, hanem csípős anyaga, a kapszaicin révén lehetővé tette a fájdalomérző idegsejtek kategorizálását, élettani, kórélettani funkcióinak és gyógyszerkutatási jelentőségének részletesebb megismerését. A klasszikus idegszabályozási elmélet szerint az érzőidegek a különféle érzeteket, fájdalmat közvetítik a bőr, az ízületek és a belső szervek felől a központi idegrendszer felé. A perifériás idegrendszer másik csoportja pedig a befutó ingerekkel kiváltott reflexek útján különféle szöveteket befolyásoló „végrehajtó”, mozgató vagy belső szerveket irányító működéseket látnak el. A kapszaicin segítségével az érző idegsejtek olyan különleges csoportját sikerült feltérképezni, amely nemcsak a klasszikus érző/fájdalomérző működéssel rendelkezik, hanem a végződéseikből közvetlenül felszabaduló peptid természetű anyagok közvetítésével a beidegzési területen gyulladásos folyamatokat
– értágulatot, duzzadást és gyulladásos sejtaktivációt – indítanak el. Mivel ezt a folyamatot idegelemek közvetítik, neurogén gyulladásnak nevezzük, amely jelentős szerepet játszik számos betegség kialakulásában, és amelyet egyetlen forgalomban lévő gyógyszercsoport sem tud megbízhatóan és hatékonyan gátolni. Pintér Erika és Szolcsányi János egy véletlen megfigyelésén alapulva munkacsoportunk felfedezte, hogy a kapszaicin-érzékeny érzőidegekből a gyulladáskeltő neuropeptideken kívül gátló hatású anyag(ok) is felszabadul(nak), a keringésbe kerül(nek) és a test távolabbi pontjain jelentősen csökkenti(k) a neurogén gyulladást és a fájdalmat. Több mint két évtizedes munkánk során bizonyítottuk, hogy ezek közül kiemelt jelentőségű a szomatosztatin nevű peptid, amely közvetíti a megfigyelt szisztémás gyulladásgátló és fájdalomcsillapító hatásokat (1. ábra). Kutatásaink fókuszában elsősorban hosszan tartó gyulladásos (pl. krónikus ízületi és légúti gyulladás) és neuropátiás fájdalomállapotok állnak, amelyek modelljeiben bizonyítottuk, hogy az érző idegekből felszabaduló szomatosztatin által közvetített ellenregulációs mechanizmus hatékony (részletek később). Gyulladás, vagy egyéb eredetű szövetkárosodás során keletkező anyagok az érzőideg-végződéseket folyamatosan aktiválják és érzékenyítik, amely tartósan fennálló, fokozott fájdalomhoz vezet. A hosszantartó aktiváció nemcsak a perifériás végződések működését változtatja meg, hanem a gerincvelő és az agy területén is átrendeződést eredményez. Kutatócsoportunk másokkal összhangban számos bizonyítékot szolgáltatott arra vonatkozóan, hogy a perifériás érzőideg-végződések gátlása hatékony gyógyszerfejlesztési célpontot jelent a tartósan fennálló fájdalomállapotok kezelésére.
A kapszaicin receptora: a TRPV1 csatorna
A TRPV1 receptor a kapszaicin-érzékeny érzőideg-végződések egyik elsőként leírt jelfogó struktúrája, amely aktiváció után Na+ és Ca2+ ionokat enged a sejtekbe, ezzel aktivációt okoz és fájdalomérzetet generál. Ezeken kívül azonban leírták már ízületi savós hártya sejtjein, bőr hámsejtjein, erek belhám és simaizom sejtjein, immunsejteken és az agyban is. A TRPV1 komplex, érzékelő funkcióval rendelkezik, a külvilágból érkező irritánsok (pl. a vanilloid szerkezetű kapszaicin) mellett fájdalmas intenzitású hő (>43°C), mechanikai és kémiai ingerek pl. gyulladás során keletkező anyagok, mint a protonok (pH<6) aktiválják, ill. érzékenyítik. Aktivátorai közül magyar vonatkozása és terápiás gyakorlatban való alkalmazása révén legismertebb a kapszaicin, emiatt kezdetben „kapszaicin receptorként” emlegették. A nagy dózisú kapszaicin gyulladás- és fájdalomcsillapító hatással rendelkezik, amelyet a TRPV1 receptoron hatva egyrészt a kapszaicin-érzékeny érzőideg-végződések átmeneti teljes funkcióképtelenné tételével, másrészt a receptor tartós, alacsony intenzitású aktivációjakor gyulladás- és fájdalomcsökkentő anyagok felszabadításával ér el. Ennek tükrében nem meglepő, hogy számos, elsősorban helyileg alkalmazható, kapszaicin-tartalmú gyógyszer, és gyógyhatású készítmény található a patikák polcain. Ezeket egy termék kivételével főleg ízületi és izomfájdalmak enyhítésére használják. A kivétel egy közelmúltban törzskönyvezett, vényköteles tapasz (Qutenza; nagy koncentrációjú, 8%-os kapszaicin), amelyet övsömörhöz és AIDS-hez társuló neuropátiás fájdalomállapotokban használnak. A kapszaicinnek köszönhetően a TRPV1 receptor a tudományos érdeklődés középpontjába került, rengeteg tanulmány született a receptor gyulladásban és fájdalomban játszott szerepére vonatkozóan. Kutatócsoportunk a receptorral nem rendelkező egerekben vizsgálta szerepét idült ízületi gyulladásos, valamint gyulladásos komponenstől mentes neuropátiás fájdalomban. A receptorhiányos állatokban az ízületi gyulladásra jellemző korai, gyulladásos és a késői, neuropátiás jellegű fájdalom is jelentősen kisebb volt, ami alátámasztotta a receptor kulcsszerepét ezekben a folyamatokban (Szabó és mtsai., 2005; Horváth és mtsai., 2018). Nem véletlen tehát, hogy a receptorral kapcsolatban rendkívül sok, intenzív gyógyszerfejlesztési projektet indítottak, ami számos TRPV1 receptort blokkoló vegyületet eredményezett. Annak ellenére azonban, hogy ezek a szájon át szedhető gátló vegyületek hatékony gyulladás- és fájdalomcsillapító hatásokat fejtettek ki számos rágcsálómodellben, sajnos még nem sikerült közülük egy gyógyszert sem törzskönyveztetni. Dollármilliárdokat költöttek ezeknek a vegyületeknek a fejlesztésére, azonban a klinikai fázisokban jelentkező mellékhatások, a testhőmérséklet nagyfokú emelkedése és az égési sérülések kockázatának növekedése miatt ezek nem kerülhettek forgalomba. Új fejlemény a vegyületek fejlesztésével kapcsolatban, hogy a közelmúltban sikerült felderíteni a hőszabályozást felborító mellékhatás pontos mechanizmusát, aminek köszönhetően új, második generációs gyógyszerjelölt szerek fejlesztése indulhatott meg.
Rokon fájdalomérző receptor: a TRPA1
A TRPA1 receptor, mint terápiás célpont a TRPV1 gátlókkal kapcsolatos kezdeti sikertelenségek után került a kutatók figyelmének középpontjába. A TRPA1 a TRPV1-hez hasonló ioncsatorna, amely ugyancsak a kapszaicin-érzékeny érzőideg-végződéseken helyezkedik el (a TRPA1-et tartalmazó érzőidegek 97%-a egyben TRPV1-pozitív is), ahol funkcionális egységet képez a TRPV1-gyel. Nem-idegi elhelyezkedése is nagyon hasonló a TRPV1-hez. Külső irritánsok (pl. mustárolaj, fahéjaldehid), fájdalmas intenzitású hideg (<17°C), mechanikai és kémiai ingerek (pl. reaktív oxigén és nitrogén gyökök, poliszulfidok, formaldehid, metilglioxál, hidrogén-peroxid) aktiválják, míg egyes gyulladáskeltő anyagok érzékenyítik. Fontos szerepe van a gyulladásban és a fájdalomban egyaránt, amelyet kutatócsoportunk is bizonyított receptor génhiányos egerekkel. Krónikus ízületi gyulladásmodellekben betöltött fájdalom- és gyulladáskeltő szerepét munkacsoportunk írta le (Horváth és mtsai., 2016), amelyre a megjelenés után néhány héttel a Nature Reviews Rheumatology kiemelt eredményként hivatkozott. Reumatoid artritiszes betegcsoporton végzett vizsgálat is megerősítette a közelmúltban a kísérletes eredményeket, fehérvérsejteken a TRPA1 mennyiség a fájdalommal, a mozgáskorlátozottsággal és a gyulladásos aktivitással is korrelált. Számos adat bizonyítja, hogy a gyulladás során a helyileg keletkező TRPA1 izgató vegyületek különböző módon képesek befolyásolni a receptor működését az érzőideg-végződéseken és a nem idegi sejteken, amely a gyulladásos kaszkád aktiválását eredményezheti. Szisztémásan, lokálisan és a központi idegrendszerbe adott TRPA1 blokkoló vegyület egyaránt csökkentette az ízületi gyulladásos fájdalmat egérmodellekben. A TRPA1 tehát a TRPV1-hez hasonlóan fontos közvetítő szerepet tölt be gyulladásban és fájdalomban, azonban nagy előnye, hogy nem vesz részt a hőszabályozásban, így gátlói nem okoznak testhőmérséklet-emelő mellékhatást. Ebből kifolyólag még ígéretesebb támadáspontja lehet az új hatásmechanizmusú fájdalom- és gyulladáscsillapító szereknek. A TRPA1 gátlók gyulladásos és idegkárosodás által kiváltott fájdalommodellekben hatékonynak és biztonságosnak bizonyultak, sőt a közelmúltban egyikük (GRC 17536, Glenmark Pharmaceuticals, Mumbai, India) sikeresen teljesített egy cukorbetegséghez társuló, idegkárosodás okozta fájdalomban szenvedő betegeken végzett klinikai vizsgálatot is.
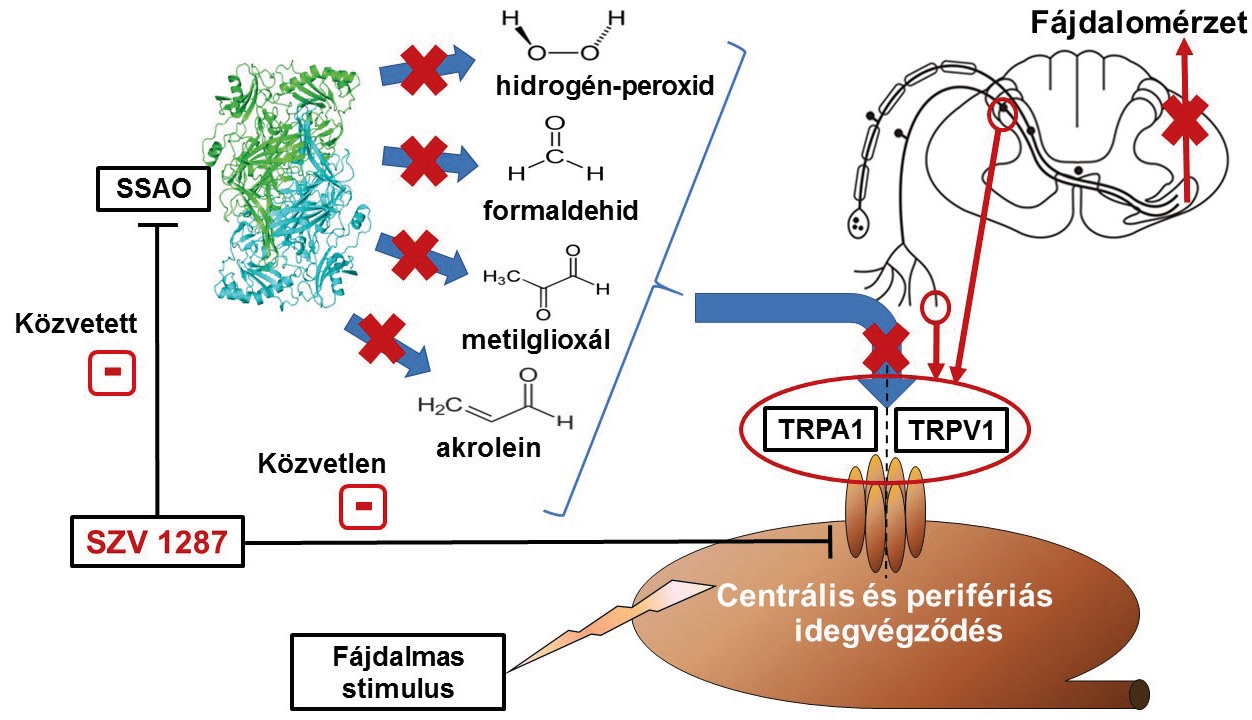
A szemikarbazid-szenzitív amin oxidáz
A TRPA1 egyes szervezeten belüli aktivátorait (hidrogén-peroxid, formaldehid, metilglioxál, akrolein) egy szemikarbazid-szenzitív amin oxidáz (SSAO) nevű enzim termeli. Ennek alapján vetődött fel bennünk, hogy érdemes megvizsgálni a TRPA1 receptor és az SSAO enzim aktivitása közötti kapcsolatot különböző eredetű és mechanizmusú fájdalomállapotokban. Alaphipotézisünk az volt, hogy az enzim gátlása következtében csökken a TRPA1-et izgató fájdalom- és gyulladáskeltő termékek keletkezése, és ez a közvetett TRPA1 aktiváció-gátláson keresztül fájdalomcsillapító hatást eredményezhet. Az SSAO az amin oxidázok családjába tartozó réztartalmú enzim, amelynek fő funkciója az élettani/kórélettani folyamatok során pl. az adrenalin és a kreatinin lebomlásakor keletkező, valamint a környezetből, pl. a kipufogógázból származó, amin típusú vegyületek átalakítása. Az enzim megtalálható szabadon a vérkeringésben, illetve sejtmembránhoz kötött formában egyaránt. Számos szövetben és szervben előfordul, azonban elsősorban a vérerek belső falának sejtjein és simaizom sejtjein, valamint zsírsejteken fejeződnek ki. Pontos élettani funkciójával ellentétben kórélettani szerepe jóval ismertebb, gyulladt területen a fehérvérsejtek érpályából való kilépésének folyamatában és az érújdonképződésben játszik jelentős szerepet. A fájdalomérzékelésben betöltött szerepét a közelmúltban kutatócsoportunk fedezte fel (Horváth és mtsai. 2016). Gyulladásban játszott fontos szerepe miatt számos betegség (pl. ízületi gyulladás, asztma, bélgyulladás) rágcsálómodelljeiben vizsgálták, és sikeresen igazolták az SSAO enzimet gátló vegyületek gyulladáscsökkentő hatását. Bár a gyulladás gyakran fájdalommal jár, ennek csillapításában való hatékonyságukat korábban nem vizsgálták, annak ellenére, hogy termékei a fájdalomérző idegvégződéseken elhelyezkedő TRPA1 receptor ismert aktivátorai. Az SSAO enzim fájdalomban betöltött szerepének leírása mellett bizonyítottuk az SSAO-gátló vegyületek fájdalomcsillapító hatását, különös tekintettel a neuropátiás fájdalomra. Ennek eredményeként szabadalmaztattuk a Semmelweis Egyetem Szerves Vegytani Intézetében Mátyus Péter csoportja által előállított új, komplex hatásmechanizmusú SSAO-gátló vegyületet, az SZV 1287-et (Helyes és mtsai., európai és USA szabadalom; US 15/303,794 & EP 14835710.6; 2016; SSAO/VAP-1 inhibitors, as novel compounds for the treatment of neuropathic pain). Az SZV 1287 egy innovatív, több támadáspontú elv szerint tervezett molekula. Egyrészt gátolja önmagában az SSAO enzimet, másrészt a szervezetben történő átalakulása során képződő vegyület révén gátolja a gyulladás kórfolyamatának egy másik kulcsenzimét, a számos gyulladás- és fájdalomkeltő vegyület termelésért felelős ciklooxigenáz enzimet is. Kutatócsoportunk emellett további támadáspontokat is felfedezett, az érző idegsejteken és az érzőideg-végződéseken a TRPA1 és TRPV1 receptorokat közvetlenül, az SSAO gátlástól függetlenül gátolja, amely jelentősen hozzájárulhat a hatékony fájdalomcsillapító hatásához (Payrits és mtsai., 2016) (2. ábra). A vegyület fájdalomcsillapító hatását különböző mechanizmusú, krónikus ízületi gyulladáshoz társuló fájdalommodellekben bizonyítottuk. Ezekben az egérmodellekben a korábban már leírt gyulladásgátló hatás mellett a referencia SSAO-gátló vegyülethez hasonlóan jelentősen csökkentette a gyulladásos fájdalmat, valamint a gyulladásos tünetek megszűnését követően fennálló, idegkárosodással magyarázható neuropátiás fájdalmat is (Horváth és mtsai., 2017 és 2018).
A gyulladás lezajlását követően megfigyelt fájdalomcsillapító hatás arra utal, hogy ez nemcsak az érzőideg-végződések, hanem a fájdalomérzékelésért felelős központi idegrendszeri területek fokozott érzékenységének gátlásából is ered. Ezt támasztja alá, hogy az ízületi gyulladásban szenvedő egerek gerincvelőjében az SZV 1287 hatékonyan csökkentette a fájdalomérzékelésért felelős területek érzékenyítésében fontos szerepet játszó immunsejtek aktivációját. Az ízületi gyulladásmodellek mellett az SZV 1287 a gyulladástól teljesen független traumás idegsérülés modellben is hatékonynak bizonyult, amellyel szintén alátámasztottuk a közvetlenül az idegi komponensekre kifejtett fájdalomcsillapító hatását (Horváth és mtsai., 2018). Az SZV 1287 fájdalom- és gyulladáscsökkentő hatásainak mechanizmusaiban részletesen vizsgáltuk továbbá a TRPA1 és TRPV1 receptorok részvételét. A TRPA1 és TRPV1 receptort aktiváló vegyületekkel kiváltott heveny gyulladás- és fájdalommodellekben az SZV 1287 csökkentette a gyorsan kialakuló, fájdalmas hőküszöb csökkenést és a fájdalomelhárító reakció időtartamát, illetve a lassan kialakuló mechanikai fájdalomküszöb csökkenést, szemben a kizárólag SSAO-gátló referenciavegyülettel, amely csak utóbbi paramétert csökkentette. Ebből arra következtethetünk, hogy az SZV 1287 fájdalomcsillapító hatásáért elsősorban az SSAO gátlás felelős, míg a közvetlen TRPA1 és TRPV1 blokkoló hatásának az érzőideg-végződések gyors érzékenyítésének gátlásában lehet szerepe. Idült ízületi gyulladás és fájdalom egérmodelljében a TRPA1 és TRPV1 receptorok SZV 1287 fájdalom- és gyulladáscsökkentő hatásában betöltött szerepét génhiányos egerek segítségével vizsgáltuk. Mivel a TRPV1 hiánya esetén az SZV 1287 nem eredményezett sem fájdalom-, sem gyulladáscsökkentő hatást, TRPA1 hiányában pedig gyulladásgátló hatást, arra következtethetünk, hogy az SZV 1287 fájdalomcsillapító hatása valószínűleg TRPV1, gyulladásgátló hatása TRPA1- és TRPV1-függő folyamat. Traumás idegsérülés egérmodelljében az SZV 1287 fájdalomcsökkentő hatása mind a TRPV1, mind a TRPA1 hiánya esetén elmaradt, ami arra utal, hogy hatásának közvetítésében mindkét receptor szerepet játszik (Horváth és mtsai., 2018). A Nemzeti Agykutatási Programban 2014-2017 között végzett kutatásaink (MTA-PTE NAP-B Krónikus Fájdalom Kutatócsoport) eredményei alapján elmondhatjuk, hogy a gyulladásos betegségek mellett az SSAO/TRPA1 ígéretes terápiás célpont ízületi gyulladáshoz és traumás károsodáshoz társuló krónikus, idegi eredetű fájdalomállapotokban, amelyre jelenleg nincs hatékony gyógyszer. Szabadalmunk fejlesztésére 2016-ban a Toxi-Coop Zrt.-vel közös 4 éves GINOP pályázatot nyertünk, melynek keretében bélben oldódó kapszula gyógyszerformát fejlesztünk neuropátiás fájdalom kezelésére. Reményeink szerint a jelenleg végzett preklinikai gyógyszerfejlesztési folyamatokban az SZV 1287 hatékonyságán kívül a hatósági előírásoknak megfelelően a biztonságosságát is bizonyítani tudjuk. Amennyiben a pályázat utolsó évében elvégzendő klinikai vizsgálatokban emberben is biztonságos lesz, akkor már nagyobb ipari partner bevonásával kezdődhet a betegeken való tesztelés. Sikeres klinikai vizsgálatok esetén az SZV 1287 új, magyar fejlesztésű vegyületként áttörést hozhat a krónikus, neuropátiás fájdalom kezelésében.

Gyulladás- és fájdalomcsökkentő – antidepresszáns, szorongás és stresszgátló mediátor
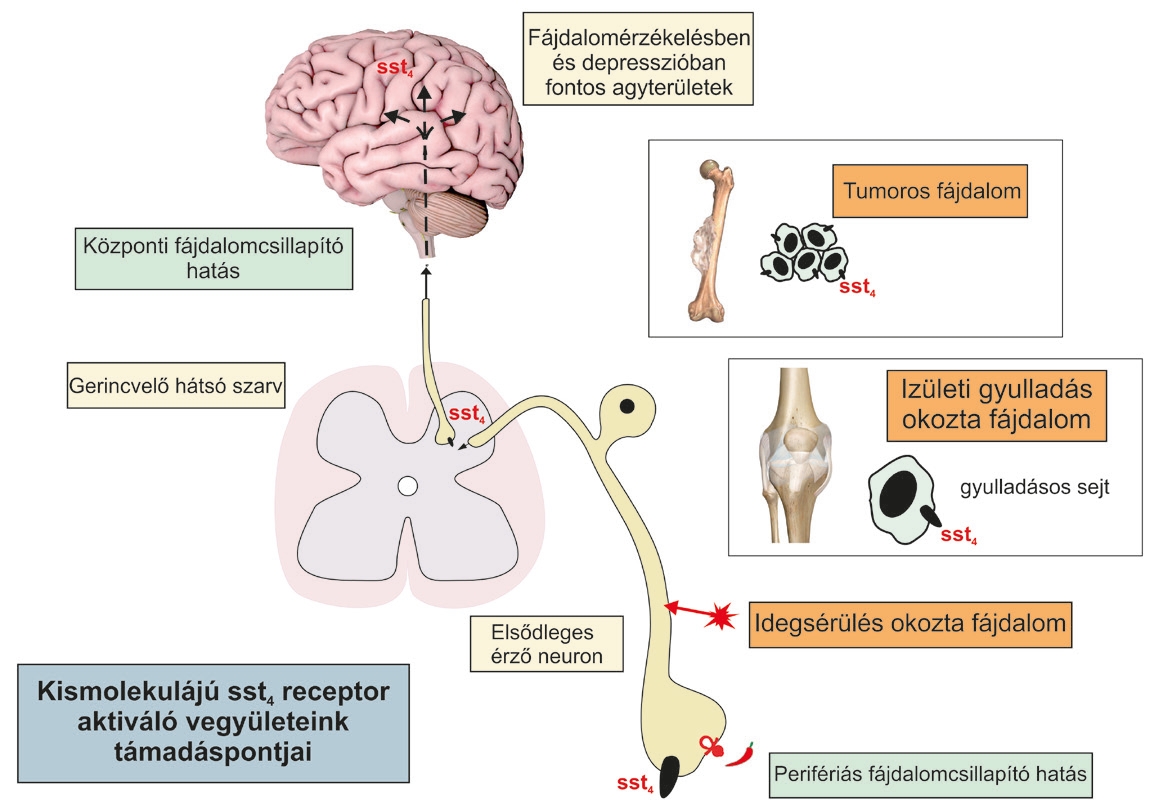
Ahogy korábban leírtuk, a kilencvenes évek közepén munkacsoportunk meglepő felfedezése volt, hogy a fájdalomérzést közvetítő kapszaicin-érzékeny, TRPV1-et és TRPA1-et kifejező érzőideg végződésekből a gyulladás- és fájdalomkeltő anyagok mellett a gyulladáscsökkentő és fájdalomcsillapító hatású szomatosztatin is felszabadul. Számos közleményben számoltunk be arról, hogy a szomatosztatin a szisztémás keringésen keresztül a test távoli pontjain is kifejti a gátló hatásait (1. ábra) (Pinter és mtsai., 2006; Szolcsanyi és mtsai., 2011). A szomatosztatin 14 és 28 aminosavból álló peptid, amelyet elsőként birkaagyból izoláltak 1972-ben, mint növekedési hormon felszabadulását gátló vegyület. Azóta kiderült, hogy a központi idegrendszerben és a periférián is számos hormon (pl. inzulin, nemi hormonok) szekrécióját gátolja. Jelenlétét a fájdalommal, hangulatszabályozással, tanulással és memóriával kapcsolatba hozható agyterületeken is kimutatták, ahol szerteágazó gátló hatásain keresztül élettani (pl. alvás-ébrenlét, mozgáskoordináció, emlékezés) és kórélettani (pl. fájdalom, depresszió, szorongás, stressz) funkciók szabályozásában vesz részt (3. ábra). Neurodegeneratív és központi idegrendszeri gyulladásos betegségekben és azok állatmodelljeiben, pl. Parkinson- és Alzheimer- kórban, epilepsziában és skizofréniában, valamint depresszióban, szorongásban és krónikus stresszben az agyi szomatosztatin szint csökken, amely arra utal, hogy ezekben az állapotokban védő funkciókkal rendelkezik. Szomatosztatin-hiányos egerekben fokozott emocionális viselkedést és depressziós betegekéhez hasonló agyi génexpressziós változásokat tapasztaltak. A kívülről agyba adott szomatosztatin egyértelműen fájdalom- és szorongáscsökkentő, valamint antidepresszáns-szerű hatásokat fejt ki. Annak ellenére, hogy a szomatosztatin széleskörű gyulladásgátló és fájdalomcsillapító hatása alapján egy új típusú, ígéretes terápiás irányt jelenthetne, rövid hatástartama és hormonháztartást befolyásoló szerteágazó egyéb hatásai miatt nem lehet erre a célra ideális gyógyszerjelölt. Szintetikus analógok azonban, amelyek hormonális mellékhatásoktól mentesek, áttörést jelenthetnek a gyulladáscsökkentő/fájdalomcsillapító gyógyszerek terén. Ehhez alapvető fontosságú az ezeket a hatásokat közvetítő célmolekuláinak azonosítása, hogy azokra a racionális gyógyszerfejlesztés elveit követve célzottan tudjunk tervezni aktiváló vegyületeket.
A szomatosztatin mint ígéretes célpont
A szomatosztatin 5 különböző, sejtmembránban elhelyezkedő célmolekuláját 1995-ben írták le (sst1-5), melyek mindegyike gátló hatásokat közvetít a célsejteken. Az sst2, sst3 és sst5 felelősek elsősorban a szomatosztatin hormontermeléseket gátló hatásaiért. Kutatócsoportunk fedezte fel, hogy ezzel szemben a gyulladáscsökkentő, fájdalomcsillapító, antidepresszáns és szorongásgátló hatásokért az sst4 receptor felelős, ezért különösen érdekes gyógyszerfejlesztési célpont lehet hormongátló hatások nélkül (Pintér és mtsai., 2006; Helyes és mtsai., 2009, Botz és mtsai., 2017) (4. ábra). Az sst4 receptor az elsődleges érzőidegsejteken, a fájdalomérző, hangulatszabályozásban és memóriafolyamatokban kulcsszerepet játszó pályarendszerek számos más területein lévő idegsejteken (gerincvelő, hippokampusz, érző agykéreg, limbikus rendszer), immunsejteken, érfali simaizom sejteken helyezkedik el. A közelmúltban specifikus módszer segítségével jellemeztük az sst4-et kifejező idegsejt-populációt ezeken az agyterületeken, és kimutattuk, hogy jól meghatározható serkentő idegsejteken található (3. ábra). E sejtek aktivációját a szomatosztatin egyértelműen az sst4-en keresztül gátolja. Az elmúlt két évtizedben számos kísérletben bizonyítottuk, hogy stabil, szintetikus szomatosztatin analógok, a hét aminosavat tartalmazó TT-232, valamint a nem-peptid szerkezetű J-2156, amelyek az sst4 receptorokat aktiválják, hatékonyan gátolják a gyulladást, a neuropátiás fájdalmat és a depressziószerű viselkedést (Helyes és mtsai., 2006, 2009, Sándor és mtsai., 2006, Scheich és mtsai. 2016, 2017a). Ezen túlmenően, irodalmi adatok szerint egy másik sst4 receptor izgató, az NNC 26-9100 protektív hatásúnak bizonyult az Alzheimer-kór és az epilepszia egérmodelljeiben. A stressz és a krónikus fájdalom klinikai, mindkét irányú összefüggéseire és közös agyi mechanizmusaira, kapcsolatrendszereire vonatkozóan számos kísérletes és klinikai adat áll rendelkezésre (Scheich és mtsai., 2017b). A hosszan tartó pszicho-szociális stressz oki tényezője és súlyosbítója lehet fájdalomállapotoknak (pl. a gyulladás vagy egyéb neurológiai eltérés nélkül testszerte jelentkező váz- és izomrendszeri fájdalomállapot, a fibromialgia, vagy a reumatoid artritisz, illetve a neuropátiás fájdalom egyéb típusai, tumoros fájdalom, stb.), valamint a krónikusan fennálló fájdalom depresszióhoz vezet. A fájdalom feldolgozásában és a hangulat szabályozásában nagyon sok közös központi idegrendszeri struktúra van, ezek komplex hálózatot képeznek, funkcionális zavaraiban számos hasonló mechanizmus játszik szerepet. A stressz- és fájdalomrendszerek kapcsolatainak a megértése, a kórélettani folyamatoknak a megismerése alapvető fontosságú olyan új gyógyszercélpontok azonosításához, amely ezekre a komplex folyamatokra egyszerre lehet hatással. A NAP-B pályázatunkban nyert eredményeink szerves folytatásaként a NAP-2 pályázat keretei között kutatócsoportunk ezen dolgozik. Mivel az sst4 receptor aktivációja egyszerre közvetít perifériás és centrális fájdalomcsillapító, gyulladásgátló, antidepresszáns és szorongásgátló hatásokat, kiváló új, egyedülálló hatás-spektrumú gyógyszerfejlesztési célmolekula (5. ábra).
Új, szabadalmaztatott sst4 aktiváló vegyületeink
A Vichem Kft. számos kismolekulájú sst4 receptort aktiváló vegyületet állított elő, amelyre hazai és nemzetközi szabadalmi beadványunk van (Waczek, Helyes és mtsai., PCT P1400432; New agents of treating neurogenic inflammation and neuropathic pain). E vegyületek sst4 receptorhoz való kötődését számítógépes modellezéssel (farmakoinformatikai kutatócsoportunkkal kollaborációban), receptoraktiváló képességét speciális sejtvonalon, hatásait rágcsálómodellekben végezzük. Jelenleg a gyógyszerfejlesztési szempontból legmegfelelőbb vezérmolekula kiválasztásán és optimalizáción dolgozunk. Négy vegyületünk szájon át adva mikrogrammos dózistartományban hatékonyan gátolja az idegsérülés okozta neuropátiás fájdalmat (80%-os fájdalomcsökkenés), továbbá szorongáscsökkentő, antidepresszáns-szerű hatásokat eredményez mozgáskoordinációt és éberséget csökkentő mellékhatás nélkül. Ez különösen fontos, mivel a jelenleg használható gyógyszerek közül egyik sem rendelkezik hasonló hatás-spektrummal. Ezek az eredmények rendkívül ígéretes új perspektívát jelenthetnek egyedülálló, kombinált fájdalomcsillapító és antidepresszáns gyógyszerek fejlesztésére.
HELYES ZSUZSANNA – HORVÁTH ÁDÁM – SZŐKE ÉVA
IRODALOM
[1] Botz B, Bölcskei K, Helyes Z.: Challenges to develop novel antiinflammatory and analgesic drugs. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017 May;9(3).
[2] Helyes Z, Pintér E, Németh J, Sándor K, Elekes K, Szabó A, Pozsgai G, Keszthelyi D, Kereskai L, Engström M, Wurster S, Szolcsányi J: Effects of the somatostatin receptor subtype 4 selective agonist J-2156 on sensory neuropeptide release and inflammatory reactions in rodents. Br J Pharmacol. 2006 149(4):405-15.
[3] Helyes Z, Pintér E, Sándor K, Elekes K, Bánvölgyi A, Keszthelyi D, Szoke E, Tóth DM, Sándor Z, Kereskai L, Pozsgai G, Allen JP, Emson PC, Markovics A, Szolcsányi J: Impaired defense mechanism against inflammation, hyperalgesia, and airway hyperreactivity in somatostatin 4 receptor gene-deleted mice. Proc Natl Acad Sci U S A. 2009 106(31):13088-93.
[4] Helyes Z, Mátyus P, Tékus V, Scheich B.: SSAO/VAP-1 inhibitors, as novel compounds for the treatment of neuropathic pain US 15/303,794 & EP14835710.6; 2016.
[5] Horvath A, Tekus V, Boros M, Pozsgai G, Botz B, Borbely E, et al. Transient receptor potential ankyrin 1 (TRPA1) receptor is involved in chronic arthritis: in vivo study using TRPA1-deficient mice. Arthritis Res Ther. 2016 Jan;18:6.
[6] Horváth Á, Menghis A, Botz B, Borbély É, Kemény Á, Tékus V, Csepregi JZ, Mócsai A, Juhász T, Zákány R, Bogdán D, Mátyus P, Keeble J, Pintér E, Helyes Z.: Analgesic and Anti-Inflammatory Effects of the Novel Semicarbazide-Sensitive Amine-Oxidase Inhibitor SzV-1287 in Chronic Arthritis Models of the Mouse. Sci Rep. 2017 Jan 9;7:39863.
[7] Horváth Á, Tékus V, Bencze N, Szentes N, Scheich B, Bölcskei K, et al. Analgesic effects of the novel semicarbazide-sensitive amine oxidase inhibitor SZV 1287 in mouse pain models with neuropathic mechanisms: involvement of transient receptor potential vanilloid 1 and ankyrin 1 receptors. Pharmacol Res. 2018 May;131:231-243.
[8] Payrits M, Sághy, Mátyus P, Czompa A, Ludmerczki R, Deme R, et al. A novel 3-(4,5-diphenyl-1,3-oxazol-2-yl)propanal oxime compound is a potent Transient Receptor Potential Ankyrin 1 and Vanilloid 1 (TRPA1 and V1) receptor antagonist. Neuroscience. 2016;324:151–62.
[9] Pintér E, Helyes Z, Szolcsányi J: Inhibitory effect of somatostatin on inflammation and nociception. Pharmacol Ther. 2006 112(2):440-56.
[10] Sándor K, Elekes K, Szabó A, Pintér E, Engström M, Wurster S, Szolcsányi J, Helyes Z: Analgesic effects of the somatostatin sst4 receptor selective agonist J-2156 in acute and chronic pain models. Eur J Pharmacol. 2006 539(1-2):71-5.
[11] Scheich B, Gaszner B, Kormos V, László K, Ádori C, Borbély É, Hajna Z, Tékus V, Bölcskei K, Ábrahám I, Pintér E, Szolcsányi J, Helyes Z: Somatostatin receptor subtype 4 activation is involved in anxiety and depression-like behavior in mouse models. Neuropharmacology. 2016 101:204-15.
[12] Scheich B, Vincze P, Szőke É, Borbély É, Hunyady Á, Szolcsányi J, Dénes Á, Környei Z, Gaszner B, Helyes Z.: Chronic stress-induced mechanical hyperalgesia is controlled by capsaicin-sensitive neurones in the mouse. Eur J Pain. 2017 Sep;21(8):1417-1431.
[13] Scheich B, Csekő K, Borbély É, Ábrahám I, Csernus V, Gaszner B, Helyes Z.: Higher susceptibility of somatostatin 4 receptor gene-deleted mice to chronic stress-induced behavioral and neuroendocrine alterations. Neuroscience. 2017 Mar 27;346:320-336.
[14] Szabo A, Helyes Z, Sandor K, Bite A, Pinter E, Nemeth J, et al. Role of transient receptor potential vanilloid 1 receptors in adjuvantinduced chronic arthritis: in vivo study using gene-deficient mice. J Pharmacol Exp Ther. 2005 Jul;314(1):111–9.
[15] Szolcsányi J, Pintér E, Helyes Z, Petho G: Inhibition of the function of TRPV1-expressing nociceptive sensory neurons by somatostatin 4 receptor agonism: mechanism and therapeutical implications. Curr Top Med Chem. 2011 11(17):2253-63.
[16] Waczek F, Helyes Zs, Őrfi L, Kéri G, Szűts T, Pintér E, Szolcsányi J, Szőke É: New agents for treating neurogenic inflammation and neuropathic hyperalgesia related disorders. PCT P1400432; 2014.
A cikk a Természet Világa 2018. decemberi (149. évf. 12. sz.) számában jelent meg.